
Cannabidiol inhibits lung cancer cell invasion and metastasis via intercellular adhesion molecule-1
This originally appeared at https://pubmed.ncbi.nlm.nih.gov/22198381/
By:
Robert Ramer,* Katharina Bublitz,* Nadine Freimuth,* Jutta Merkord,* Helga Rohde,* Maria Haustein,* Philipp Borchert,* Ellen Schmuhl,* Michael Linnebacher,† and Burkhard Hinz*,1*Institute of Toxicology and Pharmacology and †Section of Molecular Oncology and Immunotherapy, Department of General Surgery, University of Rostock, Rostock, Germany
ABSTRACT
Cannabinoids inhibit cancer cell invasion via increasing tissue inhibitor of matrix metalloproteinases-1 (TIMP-1). This study investigates the role of intercellular adhesion molecule-1 (ICAM-1) within this action. In the lung cancer cell lines A549, H358, and H460, cannabidiol (CBD; 0.001–3 mM) elicited concentration-dependent ICAM-1 up-regulation compared to vehicle via cannabinoid receptors, transient receptor potential vanilloid 1, and p42/44 mitogenactivated protein kinase. Up-regulation of ICAM-1 mRNA by CBD in A549 was 4-fold at 3 mM, with significant effects already evident at 0.01 mM. ICAM-1 induction became significant after 2 h, whereas significant TIMP-1 mRNA increases were observed only after 48 h. Inhibition of ICAM-1 by antibody or siRNA approaches reversed the anti-invasive and TIMP-1-upregulating action of CBD and the likewise ICAM-1- inducing cannabinoids D9-tetrahydrocannabinol and R(1)-methanandamide when compared to isotype or nonsilencing siRNA controls. ICAM-1-dependent antiinvasive cannabinoid effects were confirmed in primary tumor cells from a lung cancer patient. In athymic nude mice, CBD elicited a 2.6- and 3.0-fold increase of ICAM-1 and TIMP-1 protein in A549 xenografts, as compared to vehicle-treated animals, and an antimetastatic effect that was fully reversed by a neutralizing antibody against ICAM-1 [% metastatic lung nodules vs. isotype control (100%): 47.7% for CBD 1 isotype antibody and 106.6% for CBD 1 ICAM-1 antibody]. Overall, our data indicate that cannabinoids induce ICAM-1, thereby conferring TIMP-1 induction and subsequent decreased cancer cell invasiveness.—Ramer, R., Bublitz, K., Freimuth, N., Merkord, J., Rohde, H., Haustein, M., Borchert, P., Schmuhl, E., Linnebacher, M., Hinz, B. Cannabidiol inhibits lung cancer cell invasion and metastasis via intercellular adhesion molecule-1. FASEB J. 26, 1535–1548 (2012)
Besides their palliative benefits in cancer therapy, accumulating evidence suggests a potential advance of cannabinoids as anticancer agents. Accordingly, several investigations revealed antitumorigenic cannabinoid actions, such as inhibition of tumor cell proliferation (1, 2) and angiogenesis (3–5), as well as induction of apoptosis and autophagy (6–8). A possible clinical use of cannabinoids for the treatment of highly invasive cancer types is further supported by recent findings showing a decrease of tumor cell invasion by the phytocannabinoids D9-tetrahydrocannabinol (THC; refs. 9–11) and 2-[(1S,6S)-3-methyl-6-(prop-1-en-2-yl) cyclohex-2-enyl]-5-pentylbenzene-1,3-diol (CBD; cannabidiol; refs. 12–14), as well as by the hydrolysis stable anandamide analog R(1)-methanandamide (MA; ref. 11), the CB2 agonist JWH-133 (9), the endocannabinoid 2-arachidonyl glycerol (15), and the synthetic cannabinoid WIN55,212–2 (16).Among cannabinoid-based drugs, CBD has raised particular interest due to its lack of adverse psychoactive effects that limit the clinical use of classic cannabinoids. Besides its beneficial effects on inflammation, pain, and spasticity when used for the treatment of multiple sclerosis (17), CBD has been reported to exert inhibitory effects on tumor angiogenesis (18) and metastasis (12, 13, 19) and to induce cancer cell apoptosis (20, 21). Concerning its anti-invasive mechanism, one study demonstrated an inhibitor of basic helix-loop-helix transcription factors, inhibitor of differentiation-1 (Id-1), to be involved in the anti-invasive effect of CBD on breast cancer cells (12). Recently, we were able to demonstrate an anti-invasive action of CBD on human lung and cervical carcinoma cells causally linked to up-regulation of tissue inhibitor of matrix metalloproteinases-1 (TIMP-1) via a mechanism involving activation of cannabinoid receptors and transient receptor potential vanilloid 1 (TRPV1) (13). In another study, this anti-invasive pathway was likewise elicited by THC and MA (11).The present study focuses on the role of the intercellular adhesion molecule-1 (ICAM-1) in the antimetastatic action of CBD in vivo and the TIMP-1-inducing and anti-invasive action of CBD and other cannabinoids in vitro. ICAM-1, also referred to as CD54, is an inducible 80- to 110-kDa transmembrane glycoprotein belonging to the immunoglobulin superfamily. ICAM-1 is traditionally known to play a crucial role as an adhesion molecule in trafficking of inflammatory cells and in cell-to-cell interactions during antigen presentation (22). In addition, increasing evidence suggests a function for ICAM-1 in cellular signal transduction pathways by eliciting outside-in signaling (22). The presently available literature is controversial on whether upregulation or suppression of ICAM-1 expression contributes to tumor progression and metastasis. On the one hand, increasing numbers of data suggest that enhanced ICAM-1 levels on cancer cells may be involved in tumor suppression via an immuno-surveillance mechanism. Accordingly, several studies revealed increased tumor susceptibility to lymphocyte adhesion and cell-mediated cytotoxicity following transfection (23, 24) or up-regulation of ICAM-1 (25, 26) or even under conditions of basal ICAM-1 expression (27). Vice versa, down-regulation of ICAM-1 by transforming growth factor b1 has been demonstrated to decrease both lymphocyte adhesion to cancer cells as well as cancer cell cytotoxicity (28). In line with this notion, ICAM-1 expression has been reported to be negatively correlated to metastasis of several cancer types in clinical studies (29–31). On the other hand, some studies using tumor necrosis factor (TNF) as ICAM-1 inductor or ICAM-1-overexpressing cells have suggested proinvasive properties for ICAM-1 (32–34). In addition, one investigation reported that the enhanced metastatic ability of TNF-treated malignant melanoma cells is reduced by ICAM-1 antisense oligonucleotides (35).The current study was initiated on the basis of accidental findings showing a profound up-regulation of ICAM-1 in different non-small-cell lung cancer (NSCLC) cells by CBD, the above-mentioned anticarcinogenic properties of ICAM-1, and accumulating evidence pointing to a signaling function of ICAM-1 in eliciting cellular events, such as activation of mitogenactivated protein kinases (MAPKs) and activator protein-1 (AP-1) eventually leading to the production of various cytokines and adhesion molecules (22, 36–39). Consequently, an inductive action of ICAM-1 on the pivotal anti-invasive factor TIMP-1 that has likewise been shown to be induced on MAPK activation (11, 13) and to contain an AP-1 binding site in its promoter region (40, 41) appeared feasible.Here, we demonstrate for the first time that increased ICAM-1 levels elicited by CBD result in a decrease of tumor cell invasion and metastasis. ICAM-1 up-regulation by CBD was observed in a panel of lung tumor cells, lung tumor xenografts, and primary tumor cells obtained from a brain metastasis of a patient with NSCLC. The functional importance of this finding was proven by experiments showing that de novo synthesis of ICAM-1 may represent a pivotal link between upstream cannabinoid-activated receptors and receptor-elicited p42/44 MAPK activation and downstream TIMP-1-dependent inhibition of invasion. Furthermore, to the best of our knowledge, this is the first study on the anti-invasive action of cannabinoids using human primary tumor cells and the first to provide an inhibitorbased antimetastatic mechanism of a cannabinoid in an in vivo model.
MATERIALS AND METHODS
Materials(2)-CBD was purchased from Tocris (Bad Soden, Germany).N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (AM-251), (6-iodo-2-methyl-1-[2-(4-morpholinyl)ethyl]-1H-indol-3-yl) (4-methoxyphenyl)methanone (AM-630), capsazepine, PD98059, and SB203580were bought from Alexis Deutschland (Gru¨nberg, Germany).Dulbecco’s modified Eagle’s medium (DMEM) with 4 mMl-glutamine and 4.5 mg/ml glucose was from Cambrex BioScience Verviers S.p.r.l. (Verviers, Belgium). Fetal calf serum(FCS) and penicillin-streptomycin were obtained from PANBiotech (Aidenbach, Germany) and Invitrogen (Karlsruhe,Germany), respectively. Dimethyl sulfoxide (DMSO), 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES),NaCl, ethylenediaminetetraacetic acid (EDTA), Triton X-100,and glycerol were bought from Applichem (Darmstadt, Germany). Phenylmethylsulfonyl fluoride (PMSF), leupeptin,and aprotinin were obtained from Sigma (Taufkirchen, Germany). Isotype control and neutralizing ICAM-1 antibodiesfor in vitro and in vivo experiments were purchased from R&DSystems (Minneapolis, MN, USA).
Cell culture
A549, H358, and H460 cells were maintained in DMEM with 4 mM l-glutamine and 4.5 mg/ml glucose supplemented with 10% heat-inactivated FCS, 100 U/ml penicillin, and 100 mg/ml streptomycin. Cells were grown in a humidified incubator at 37°C and 5% CO2. All incubations were performed in serum-free medium. Phosphate-buffered saline (PBS) was used as a vehicle for the tested substances with a final concentration of 0.1% (v/v) ethanol (for all cannabinoids) or 0.1% (v/v) DMSO (for AM-251, AM-630, capsazepine, PD98059, and SB203580). Primary metastasizing lung tumor cells were obtained from resections of brain metastases of 2 patients with NSCLC (see Fig. 8 and Table 2). Samples from brain metastasis of lung carcinoma were excised, stored at 4°C in PBS, and immediately transferred to the laboratory. Samples were minced, and single-cell suspensions were generated. Data for patient 1 (see Fig. 8 and Table 2) were obtained from experiments with cells from a resection of a brain metastasis of a 67-yr-old male Caucasian with NSCLC. For these experiments, cells were passaged 5–7 times without intermediate freezing steps in DMEM containing 20% FCS and 100 U/ml penicillin and 100 mg/ml streptomycin. Experiments were performed using cells from passage 5–7 in serum-free DMEM after cells reached ;70% confluence. Results for patient 2 (see Table 2) were obtained from Matrigel invasion assays using cells from a brain metastasis of a 47-yr-old female Caucasian with NSCLC at passage 0. All patients were informed about the establishment of cellular models from their individual tumors and gave informed consent in written form. The procedure was approved by the institutional ethics committee at the University of Rostock.
Matrigel invasion assay
The invasiveness of cells was quantified using a modified Boyden chamber technique with Matrigel-coated membranes (BD Biosciences, Oxford, UK), according to the manufacturer’s instructions, as described recently (11, 13, 42). In this assay, cells must overcome a reconstituted basement membrane by proteolytic degradation of a Matrigel layer and active migration. In brief, the upper sides of the transwell inserts (8-mm pore size) were coated with 28.4 mg Matrigel/ insert in a 24-well plate format. Cells were used at a final concentration of 5 3 105 cells/well in a volume of 500 ml serum-free DMEM in each insert and treated with test substances or vehicles for the indicated times. DMEM containing 10% FCS was used as a chemoattractant in the companion plate. Following incubation in a humidified incubator at 37°C and 5% CO2 for the indicated times, the noninvading cells on the upper surface of the inserts were removed with a cotton swab, and viability of invaded cells on the lower surface was measured by the colorimetric 4-[3-(4-iodophenyl)-2-(4-nitrophenyl)-2H-5-tetrazolio]-1,6-benzene disulfonate (WST-1) test (Roche Diagnostics, Mannheim, Germany). For calculation of migration, the viability of cells on the lower side of uncoated invasion chambers was determined by the WST-1 test. Invasion was expressed as the invasion index, which is calculated as the absorbance at 490 nm of cells that invaded through Matrigel-coated Boyden chambers divided by absorbance of cells that migrated through uncoated control inserts with equal treatment [(invasion/migration)3100%].
Quantitative reverse transcriptase-polymerase chain reaction (RT-PCR) analysis
Cells were seeded into 24-well plates at a density of 1 3 105 cells/well. Following incubation with the respective cannabinoid or its vehicle for the indicated times, cell culture media were removed, and cells were lysed for subsequent RNA isolation using the RNeasy total RNA Kit (Qiagen, Hilden, Germany). b-Actin (internal standard), ICAM-1, and TIMP-1 mRNA levels were determined by quantitative real-time RTPCR, as described recently (43). Primers and probes for human b-actin, ICAM-1, and TIMP-1 were TaqMan Gene Expression Assays (Applied Biosystems, Darmstadt, Germany).
Western blot analysis
For determination of TIMP-1 protein levels (see Fig. 3), cells grown to confluence in 24-well-plates were incubated with test substances or vehicles. Afterward, cell culture media were centrifuged at 500 g and used for Western blot analysis. For all other blots, TIMP-1 was determined in cell culture media collected from the upper Boyden chambers at the end of the respective invasion experiment. Total protein in the cell culture medium was measured using the bicinchoninic acid assay (Pierce, Rockford, IL, USA). For analysis of ICAM-1 expression, cells grown in 24-well plates (see Figs. 1A, 4, and 6) or 6-well-plates (see Figs. 1B, C, 2, and 7) were incubated with test substances or vehicles. For Western blot analysis of p42/44 and phospho-p42/44 MAPK, cells grown to confluence in 6-well-plates were incubated with test substances or vehicle for the indicated times. In case of lysate analyses, cells were washed, harvested, lysed in solubilization buffer (50 mM HEPES, pH 7.4; 150 mM NaCl; 1 mM EDTA; 1% (v/v) TritonX-100; 10% (v/v) glycerol; 1 mM PMSF; 1 mg/ml leupeptin; and 10 mg/ml aprotinin), homogenized by sonication, and centrifuged at 10,000 g for 5 min. Supernatants were used for Western blot analysis.All proteins were separated on a 10% sodium dodecyl sulfate-polyacrylamide gel (Applichem). Following transfer to nitrocellulose and blocking of the membranes with 5% milk powder, blots were probed with specific antibodies raised to TIMP-1 (Oncogene Research Products, San Diego, CA, USA), ICAM-1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), p42/44 MAPK, or phospho-p42/44 MAPK (Cell Signaling Technology, Danvers, MA, USA). Membranes were probed with horseradish peroxidase-conjugated Fab-specific antimouse IgG for detection of TIMP-1 and ICAM-1 (New England BioLabs, Frankfurt, Germany) or anti-rabbit IgG (Cell Signaling Technology) for analysis of p42/44 MAPK activation. Densitometric analysis of band intensities was achieved by optical scanning and quantifying using the Quantity One 1-D Analysis Software (Bio-Rad, Munich, Germany). Vehicle controls were defined as 100% for evaluation of changes in protein expression. To ensure that equal amounts of protein in cell culture medium used for protein analysis of TIMP-1 had been transferred to the membrane, proteins on Western blot membranes were stained with Ponceau Red (Carl Roth, Karlsruhe, Germany). To ascertain equal protein loading in Western blots of cell culture medium, a band with a size of ;65 kDa that appeared unregulated is shown as a loading control for protein analysis of cell culture medium. To ascertain equal protein loading in Western blots of cell lysates, membranes were probed with an antibody raised to b-actin (Calbiochem, Bad Soden, Germany)
siRNA transfections
Cells were transfected with small interfering RNA (siRNA) targeting the indicated sequence of ICAM-1 using RNAiFect as transfection reagent (Qiagen) or nonsilencing negative control RNA (Eurogentec, Seraing, Belgium). The target sequence of the ICAM-1 siRNA (Qiagen) was 59-CGGCCAGCTTATACACAAGAA-39. A BLAST search revealed that the sequence selected did not show any homology to other known human genes. Transfections were performed according to the manufacturer’s instructions. For invasion assays, cells grown to confluence were transfected with 1.25 mg/ml siRNA or nonsilencing siRNA as negative control with an equal ratio (w/v) of RNA to transfection reagent for 24 h in medium supplemented with 10% FCS. Subsequently, cells were trypsinized, centrifuged at 200 g, resuspended to a final density of 5 3 105 cells in 500 ml of serum-free DMEM containing the same amounts of siRNA or nonsilencing siRNA to provide constant transfection conditions, and seeded for invasion analysis, as described above.
Induction of A549 xenografts in nude mice
All animal experiments were approved by the Animal Ethics Committee at the University of Rostock. Tumors were induced in NMRI mice (nu/nu; Charles River Laboratories, Frederick, MD, USA) by subcutaneous flank inoculation of 1 3 107 A549 lung tumor cells. Animals were assigned randomly to a vehicle and a CBD group and were injected intraperitoneally every 72 h with vehicle or CBD (5 mg/kg body wt). The treatment started 7 d after subcutaneous injection of tumor cells into the dorsal right side. After 29 d, animals were sacrificed, and tumors were explanted for protein analysis. Therefore, tissue parts were quick-frozen in liquid nitrogen. For Western blot analysis, homogenates were treated as described previously (14).
Mouse model of tumor metastasis
Athymic nude mice (NMRI-nu/nu; Charles River Laboratories) were given injections of A549 cells (13106 cells/100 ml in PBS) through the lateral tail vein (d 1) and, after 24 h(d 2), were treated intraperitoneally with CBD (5 mg/kg body wt), vehicle, isotype control, or ICAM-1 antibody. Treatment protocols for the evaluation of the antimetastatic action of CBD in vivo revealed 5 mg/kg CBD as an appropriate dose for this purpose (13). CBD or its vehicle was administered every 72 h. Isotype control and ICAM-1 antibodies were used at a dose of 5 mg/mouse and were administered 13/wk. Mice were sacrificed on d 28, and total lungs were evaluated for metastases nodules. To contrast lung nodules, lungs were fixed in Bouin’s fluid (15 ml saturated picrinic acid, 5 ml formaldehyde, and 1 ml glacial acetic acid), and metastatic nodes were scored under a stereoscopic microscope in an investigator-blinded fashion. For histopathological examination, lung samples were fixed in 4% formalin. Paraffin sections were stained with hematoxylin and eosin. All experimental protocols for animal experiments were conducted in accordance with the policies of the Animal Ethics Committee at the University of Rostock.
Statistical analysis
Invasion indices and, mRNA and protein levels are indicated as means 6 se. Numbers of metastatic nodules are shown as box plots (whiskers: 5–95 percentile). Comparisons between groups were performed with Student’s 2-tailed t test or with 1-way ANOVA plus Bonferroni test using GraphPad Prism 5.00 (GraphPad Software, San Diego, CA, USA). Results were considered to be statistically significant at values of P , 0.05.
RESULTS
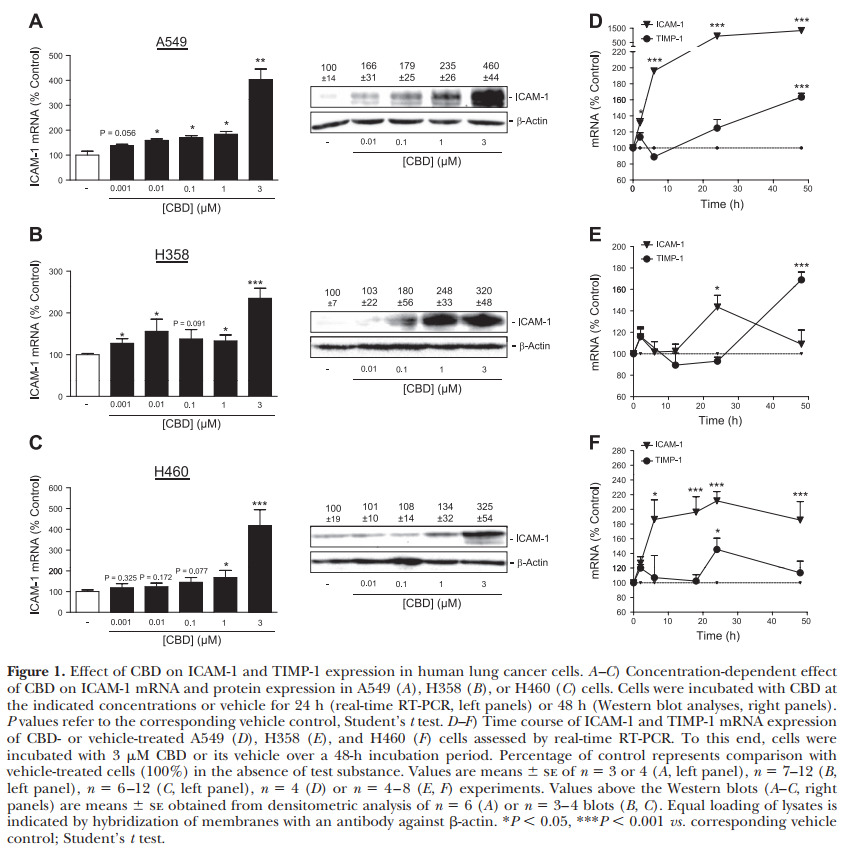
Concentration dependence of CBD’s action on ICAM- 1 mRNA and protein expression
Treatment of A549, H358, and H460 cells with CBD caused a concentration-dependent increase of ICAM-1 mRNA levels following a 24-h incubation period (Fig. 1A–C, left panels) that was significant at concentrations as low as 0.01 mM in A549 (Fig. 1A, left panel), 0.001 in H358 (Fig. 1B, left panel) and 1 mM in H460 cells (Fig. 1C, left panel). Western blot analyses revealed a corresponding concentration-dependent ICAM-1 protein up-regulation by CBD in all three cell lines tested after a 48-h incubation (Fig. 1A–C, right panels).
Time course of the stimulatory effect of CBD on ICAM-1 and TIMP-1 mRNA expression
In experiments addressing the effect of 3 mM CBD on the expression of ICAM-1 and TIMP-1 mRNA, CBD exhibited a time-dependent induction of ICAM-1 mRNA that became significant following a 2-h incubation of A549 (Fig. 1D), a 24-h incubation of H358 (Fig. 1E), and a 6-h incubation of H460 cells (Fig. 1F). TIMP-1 mRNA up-regulation by CBD became first evident after a 48-h incubation period of A549 and H358, or a 24-h incubation of H460 cells (Fig. 1D–F). Thus, in all 3 cell lines, ICAM-1 up-regulation by CBD appeared prior to TIMP-1 induction.
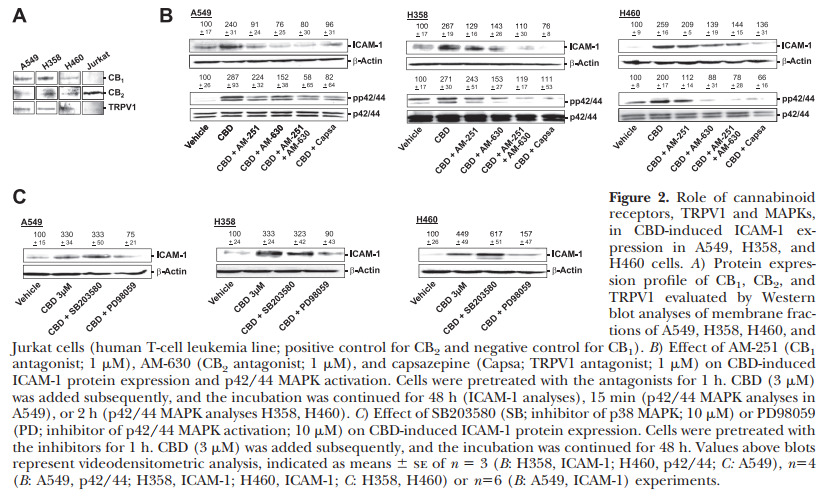
Involvement of cannabinoid receptors, TRPV1, and MAPKs, in CBD-induced ICAM-1 expression
Recently, we established that cannabinoid receptors 1 and 2 (CB1 and CB2), as well as TRPV1, confer the antiinvasive effect of CBD in A549, H358, and H460 cells (13, 14). In a first set of experiments, the expression of these receptors was confirmed by Western blot analyses of membrane fractions of A549, H358, and H460 cells (Fig. 2A). To investigate whether CBD’s ICAM-1-inductive action shares the same upstream targets, we evaluated the impact of antagonists to CB1 (AM-251), CB2 (AM-630) and TRPV1 (capsazepine) on CBD-induced ICAM-1 expression. As expected, antagonists to both cannabinoid receptors and TRPV1 suppressed CBD-induced ICAM-1. All antagonists were used at a concentration of 1 mM, which has been reported to be within the range of concentrations inhibiting CB1-, CB2- and TRPV1-dependent events (1, 11, 13, 14, 44).For potential downstream targets of receptor activation, we further addressed the influence of p42/44 and p38 MAPK inhibitors on ICAM-1 expression by CBD. In all cell lines tested, the CBD-induced ICAM-1 expression was reversed by the inhibitor of p42/44 MAPK activation, PD98059, whereas the inhibitor of p38 activity, SB203580, left the CBD-induced ICAM-1 expression virtually unaltered (Fig. 2C). As further shown, antagonists to cannabinoid receptors and TRPV1 diminished CBD-induced p42/44 MAPK activation to an extent comparable to its inhibitory action on CBD-induced ICAM-1 expression (Fig. 2B; bottom panels), thus confirming the role of p42/44 MAPK as a causal link between receptor activation and ICAM-1 induction by CBD.
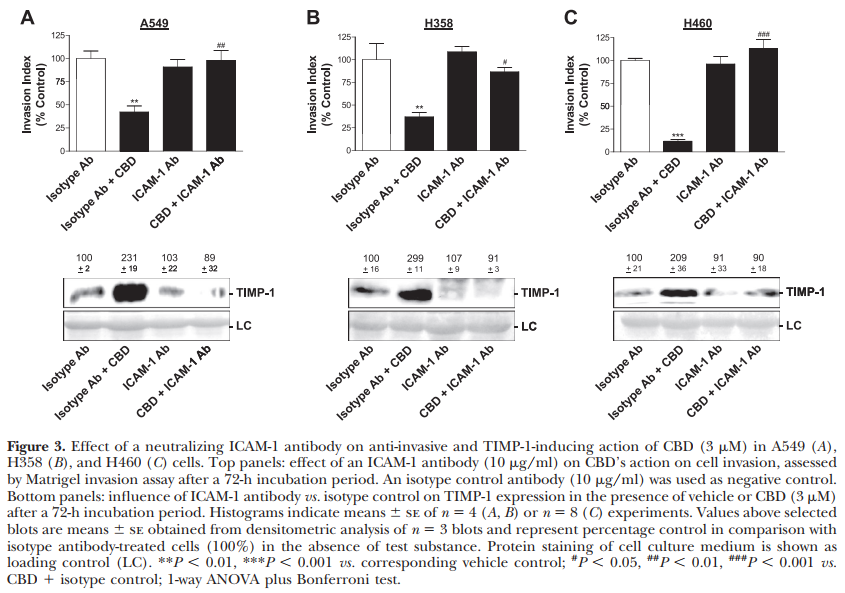
Reversal of CBD-induced TIMP-1 expression and decrease of invasiveness by a neutralizing ICAM-1 antibody
To investigate a causal link between the CBD-induced ICAM-1 and TIMP-1 up-regulation and the concomitant inhibition of invasion by CBD, a neutralizing antibody to ICAM-1 was tested for its inhibitory action on CBDinduced TIMP-1 expression and inhibition of invasiveness in A549, H358, and H460 cells. In all cell lines investigated, the ICAM-1 antibody was shown to reverse the CBD-induced TIMP-1 expression as compared to cells treated with isotype control antibody (Fig. 3A–C, bottom panels). Both ICAM-1 antibody and isotype control antibody were likewise tested for their influence on decreased cancer cell invasiveness induced by CBD. Again, in all cell lines, the ICAM-1 antibody inhibited CBD’s anti-invasive action (Fig. 3A–C, top panels)
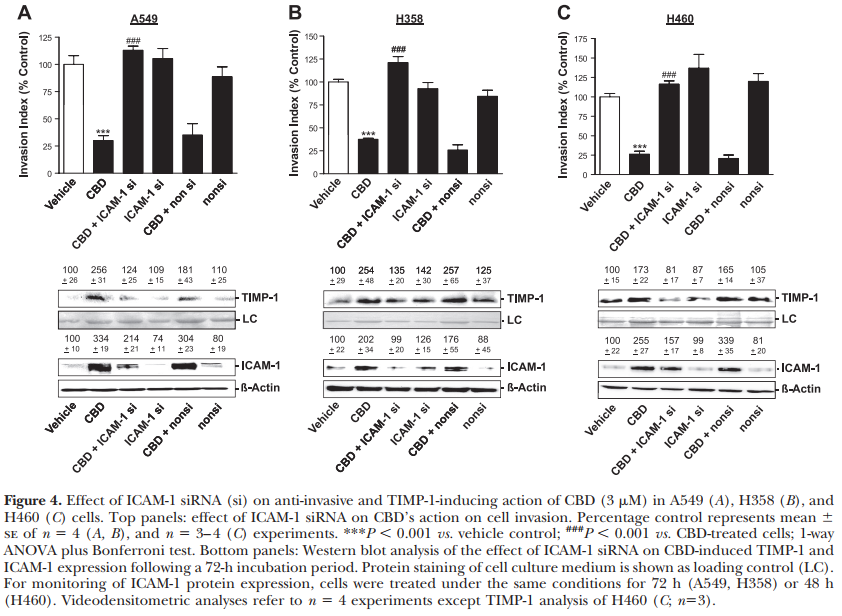
Reversal of CBD-induced TIMP-1 expression and decrease of invasiveness by ICAM-1 knockdown
To further confirm a causal link between the CBDmediated ICAM-1 and the subsequent TIMP-1-dependent inhibition of invasion, the expression of ICAM-1 was blocked by transfecting cells with ICAM-1 siRNA. Using 1.25 mg/ml siRNA targeting the ICAM-1 sequence and a nonsilencing control siRNA, transfections of A549, H358, and H460 cells were performed in Boyden Chamber assays. As indicated in Fig. 4, Western blot analyses demonstrated transfection of the respective cell line with ICAM-1 siRNA to be sufficient to decrease CBD-induced ICAM-1 expression without alteration of the basal expression of ICAM-1. As a consequence of ICAM-1 siRNA-elicited decrease of CBDinduced ICAM-1 expression, the CBD-induced TIMP-1 expression was found to be virtually abolished. Thus, abrogation of ICAM-1 expression using the siRNA approach also conferred an inhibition of CBD’s antiinvasive properties in all cell lines tested without interference with basal invasiveness (Fig. 4A–C, top panels).
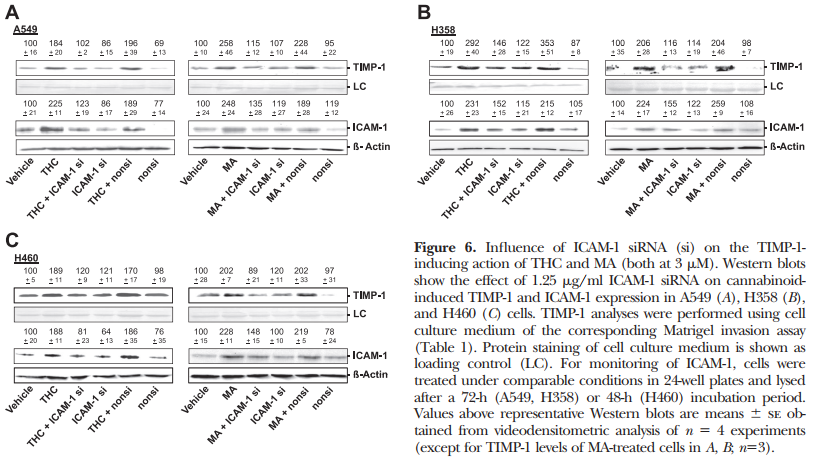
Reversal of THC- and MA-induced TIMP-1 expression and decrease of invasiveness by ICAM-1 knockdown and ICAM-1 antibody
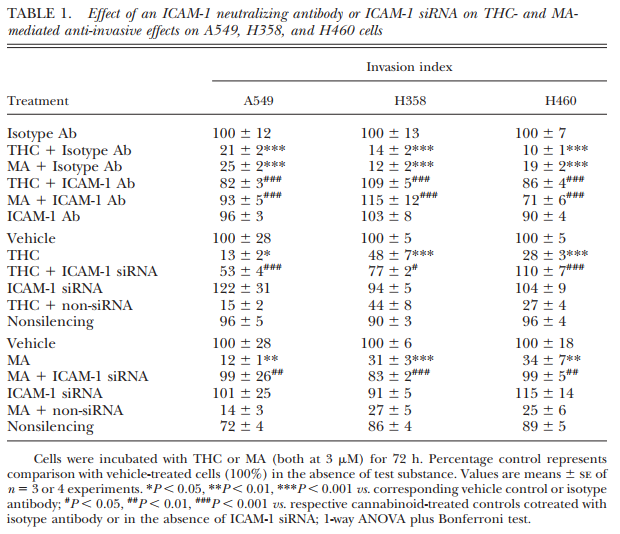
As recently shown by our group, TIMP-1 up-regulation is likewise a key player of the anti-invasive action of MA and THC in cervical carcinoma (HeLa, C33A) and A549 lung tumor cells (11). To exclude the possibility that the crucial role of ICAM-1 in the signaling cascade conferring TIMP-1 induction and subsequent inhibition of invasion is limited to CBD only, we performed further experiments with MA and THC using the same experimental settings. Real-time RT-PCR analyses revealed an up-regulation of ICAM-1 mRNA following a 24-h incubation with 3 mM MA and THC as compared to vehicle controls in all cell lines tested (Fig. 5A). To confirm the data obtained from CBD-treated tumor cells, we further investigated a causal link between MAand THC-induced ICAM-1 and TIMP-1 expression. These experiments revealed an inhibitory action of the ICAM-1 neutralizing antibody on cannabinoid-induced TIMP-1 expression. These latter findings were further substantiated by RNA interference targeting ICAM-1. As expected, in these experiments, 1.25 mg/ml ICAM-1 siRNA elicited a reduction of TIMP-1 up-regulation by MA and THC, as depicted in representative blots in Fig. 6. Table 1 provides an overview on the involvement of ICAM-1 in the anti-invasive impact of MA and THC on A549, H358, and H460 cells. The data presented here demonstrate that the ICAM-1 neutralizing antibody and knockdown of ICAM-1 expression by transfection with ICAM-1 siRNA confer a significant abrogation of cannabinoid-mediated decrease of invasion compared to the respective controls.
Effect of cannabinoids on the invasiveness of primary NSCLC cells
To obtain additional support for the anti-invasive action of cannabinoids, we analyzed tumor cells obtained from surgeries of patients with lung tumors. Biopsies were taken from brain metastases of one male and one female patient, respectively (Table 2). After biopsies were taken, NSCLC diagnosis was confirmed. For further analysis of the involvement of ICAM-1 and TIMP-1 in a possible anti-invasive action of cannabinoids, cells from patient 1 that exerted a profound ex vivo proliferation in cell culture flasks were expanded to passages 5–7 in order to provide a sufficient amount of cells before analysis of invasion. Cells from patient 2 were subjected to the Boyden chambers without intermediate expansion. As indicated in Table 2, all cannabinoids tested exerted significant anti-invasive effects on primary lung tumor cells at a final concentration of 3 mM as compared to the respective vehicle controls. The migration through uncoated Boyden chambers and the viability was left virtually unaltered by all cannabinoids tested under the high-cell-density conditions of the Matrigel invasion assay (53105 cells in a volume of 500 ml/well of a 48-well plate; data not shown).

Effect of cannabinoid-induced ICAM-1 on TIMP-1 expression and invasiveness of primary NSCLC cells
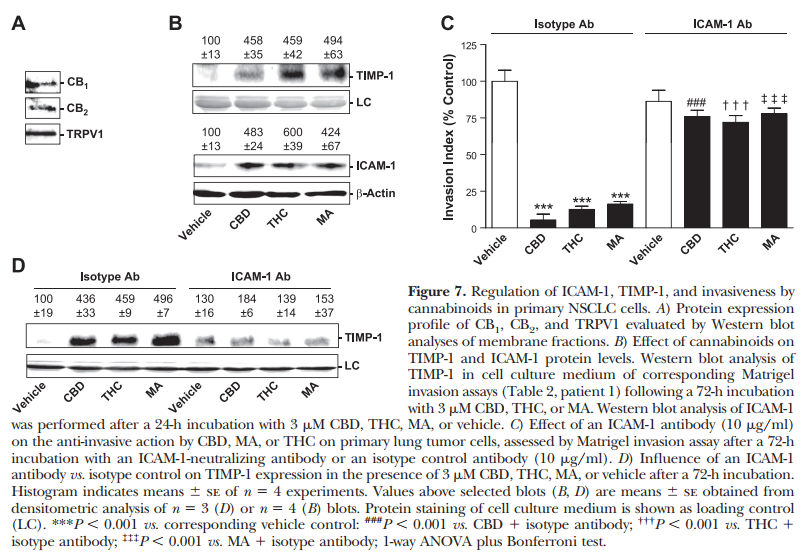
For further key experiments performed to confirm the mode of action of cannabinoids on tumor cell invasion, we used the primary lung tumor cells from patient 1. Data presented in Fig. 7 refer to experiments obtained from passages 5–7 of tumor cells from patient 1.To characterize the expression profile of cannabinoid-activated receptors on primary cells, membrane fractions were analyzed in Western blot experiments. As shown in Fig. 7A, both cannabinoid receptor subtypes, as well as TRPV1, were detected.In line with our findings in A549, H358, and H460 cells, the sequential up-regulation of ICAM-1 and TIMP-1, with ICAM-1 appearing prior to TIMP-1, was also detected in primary lung tumor cells. Accordingly, real-time RT-PCR analysis revealed induction of ICAM-1 mRNA following a 4-h incubation period with 3 mM CBD [17268% vs. vehicle control (100613%), P,0.01, Student’s t test], whereas TIMP-1 mRNA expression was left virtually unaltered [11368% vs. vehicle control (100610%), P50.3383, Student’s t test]. Following a 24-h incubation, however, an induction of both ICAM-1 [673649% vs. vehicle control (10068%), P,0.001, Student’s t test] and TIMP-1 [159614% vs. vehicle control (10065%), P,0.01, Student’s t test] was observed in the presence of 3 mM CBD. In addition, Western blot analysis of cell culture medium obtained from Boyden chamber assays and cell lysates revealed an up-regulation of TIMP-1 and ICAM-1 protein by 3 mM CBD, THC, and MA (Fig. 7B).To further confirm an involvement of ICAM-1 in the anti-invasive effect of cannabinoids, we tested the impact of equimolar concentrations of CBD, THC, and MA on the invasive behavior of primary lung tumor cells. Similar to the findings obtained from experiments with lung tumor cell lines, coincubation of primary lung tumor cells with the neutralizing antibody against ICAM-1 reversed the cannabinoid-induced TIMP-1 expression and the subsequent inhibition of invasiveness (Fig. 7C, D).
In vivo investigations of a functional implication of ICAM-1 in the antimetastatic effect of CBD
To investigate a potential CBD-induced ICAM-1 and TIMP-1 up-regulation in vivo, athymic nude mice were xenografted with A549 cells, randomized into 2 groups, and treated with vehicle or CBD (5 mg/kg, every 72 h) over 28 d. Tumor sizes of 4 animals/experimental group measured after a 28-d treatment with vehicle or CBD revealed a mean diameter 6 se of 1405 6 273 mm3 in vehicle-treated animals and 416 6 125 mm3 in CBDtreated animals (P50.0165, Student’s t test). As shown in Fig. 8A, CBD caused a profound induction of ICAM-1 and TIMP-1 protein assessed by Western blots of tumor tissues.In a metastasis model, CBD was found to significantly inhibit lung metastatic nodules. This inhibition was completely reversed by a neutralizing antibody to ICAM-1. Although the ICAM-1 antibody alone also tended to inhibit the number of metastatic nodules, the effect was not significant (isotype vs. ICAM-1 antibodytreated animals: P50.1154).
DISCUSSION
Cannabinoids are currently discussed as tools for new anticancer therapies. Besides a great body of experimental evidence pointing to an antitumorigenic action by these compounds, this notion is particularly supported by the lack of severe adverse side effects of cannabinoids as compared to the generalized toxic actions of conventional chemotherapies. Furthermore, several substantial side effects of chemotherapeutics, such as emesis and collateral toxicity on noncancerous tissues, have been demonstrated to be even attenuated on treatment with cannabinoids (45). Within cannabinoid-based substances, the phytocannabinoid CBD has emerged as a particularly interesting drug due to its lack of adverse psychoactive effects, as well as its considerable antitumorigenic properties (45).
Using different human lung cancer cell lines, primary tumor cells, and an in vivo intravenous metastasis model, we performed the present study to analyze the contribution of ICAM-1 to the anti-invasive action of CBD that has recently been described to be mediated via cannabinoid receptor-, TRPV1-, and MAPK-dependent up-regulation of TIMP-1 in human cervical and lung cancer cells (13).Analyses of mRNA and protein expression revealed aconcentration- and time-dependent increase of ICAM-1, with significant effects at CBD concentrations as low as 0.01, 0.001, and 1 mM for A549, H358, and H460 cells, respectively. Given that mean CBD plasma concentrations of 0.036 mM are achieved in patients undergoing a 6-wk oral treatment with CBD at doses of 10 mg/kg/d (46), the ICAM-1-inductive action of CBD occurred approximately in a range of therapeutically relevant concentrations, at least in A549 and H358 cells. In addition, ICAM-1 induction observed in A549 cells correlated with the recently established anti-invasive effect of CBD on A549 lung tumor cells that was likewise detectable at CBD concentrations of 0.01 mM and significant at 0.1 mM (13).There are several lines of evidence supporting a role of ICAM-1 as a functional link between cannabinoid receptor- and TRPV1-elicited p42/44 MAPK activation and downstream TIMP-1-dependent inhibition of invasion. First, antagonists to CB1 and CB2, as well as an antagonist to TRPV1, were shown to inhibit CBDinduced ICAM-1 expression, which is in line with CBD’s recently established cannabinoid- and TRPV1-dependent up-regulation of TIMP-1 expression (13) and subsequent anti-invasive action (13, 14). Second, CBDinduced ICAM-1 expression was found to occur via a receptor-dependent activation of p42/44 MAPK—a pathway likewise shared by the recently published TIMP-1-dependent anti-invasive action by this cannabinoid (13).Third, in all cell lines, as well as primary tumor cells
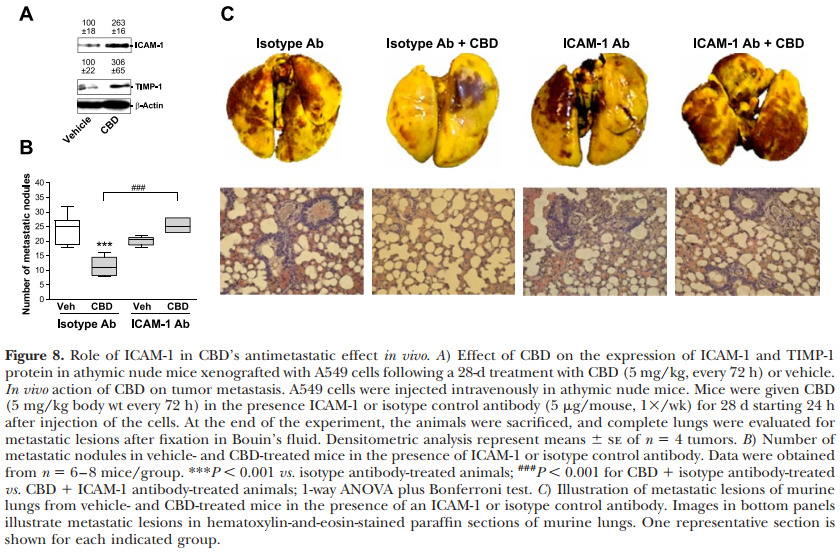
from a brain metastasis of a patient with NSCLC, CBD elicited a sequential up-regulation of ICAM-1 and TIMP-1 mRNA. Although up-regulation of both mRNAs displayed a high cell-dependent variation between 2 and 24 h for ICAM-1 and 24 to 48 h for TIMP-1, ICAM-1 was induced by CBD prior to TIMP-1 in either case. Fourth, both neutralization of ICAM-1 using a specific antibody and post-transcriptional knock-down of ICAM-1 using the siRNA approach were found to elicit a comparable inhibition of TIMP-1 induction and of the decreased invasiveness by CBD. Finally, and most important, up-regulation of both anti-invasive mediators, ICAM-1 and TIMP-1, by CBD was substantiated in vivo by use of thymic-aplastic nude mice xenografted with A549 cells. A functional involvement of ICAM-1 in the suppression of metastasis by CBD in vivo was proven by experiments showing that a neutralizing antibody to ICAM-1 led to a complete reversal of the antimetastatic effect of CBD.The possibility that an ICAM-1/TIMP-1-dependent anti-invasive action is restricted to CBD could be excluded by investigating 2 additional cannabinoids. In key experiments with the phytocannabinoid THC and the stable anandamide analog MA, neutralization or knockdown of cannabinoid-induced ICAM-1 was again found to suppress cannabinoid-elicited TIMP-1 induction and decreased cancer cell invasiveness. Finally, an ICAM-1/TIMP-1-dependent anti-invasive action elicited by the 3 cannabinoids was also confirmed in primary tumor cells obtained from a brain metastasis of apatient with NSCLC, supporting a clinical implication of our findings.A deviation between the recently reported TIMP-1- dependent anti-invasive action of CBD and its effect on ICAM-1 is the lack of inhibition of CBD-induced ICAM-1 expression by the p38 MAPK inhibitor SB203580. On the basis of these data, it is tempting to speculate that, in contrast to p42/44 MAPK activation, the induction of the p38 MAPK pathway may represent an additional, ICAM-1-bypassing effect of cannabinoids to up-regulate TIMP-1. Alternative mechanisms may lie in a direct activation of p38 MAPK by ICAM-1 (47) or an ICAM-1 ectodomain shedding (48) with a subsequent p38 MAPK induction by a soluble truncated ICAM-1 (49).In view of the fact that both TIMP-1 induction and decreased invasiveness by cannabinoids were abolished by a monoclonal human ICAM-1 antibody that reacts with ICAM-1 expressed on the surface of cells, it is tempting to speculate that the integral membrane glycoprotein ICAM-1 confers an outside-in signaling following up-regulation through cannabinoids. Although we did not use classical experimental settings (i.e., cross-linking antibodies or proteins, engagement with fibronectin) to simulate interaction of ICAM-1 with its ligand, b2 integrin molecules, binding of components of the extracellular matrix available in Matrigel to ICAM-1 could elicit cellular activation that eventually leads to increased TIMP-1 expression. However, given the fact that TIMP-1 was also induced on cannabinoid treatment with cells in Matrigel-free systems, other mechanisms appear feasible. Interestingly, there are several other studies that did not use cross-linking or ligation strategies, but were likewise able to show a contribution of de novo synthesized ICAM-1 on the subsequent invasiveness of A549 (32, 34) or breast cancer cells (33), on adhesion of A549 cells (50), or on migration of endothelial cells (51) with all responses being sensitive to inhibition by specific ICAM-1 antibodies. However, the present study is in line with previous reports showing that ICAM-1 can initiate phosphorylation of p42/44 MAPK, activation of AP-1, and production of cytokines (e.g., interleukin-1, interleukin-8, RANTES) or adhesion molecules (vascular cell adhesion molecule-1) (36–39). Given the fact that the TIMP-1-promotor region also contains an AP-1 binding site (40, 41), the findings reported here appear plausible in context with the literature. Our results are in agreement with numerous studies suggesting ICAM-1 expression as a beneficial factor for patient prognosis, tumor size, relapse of cancer, and metastasis (28–31, 52–54). However, conflicting data published by others need to be addressed here. In particular, there are some findings implying a proinvasive function of ICAM-1 on tumor cells (32–34). This apparent discrepancy may be explained by the different experimental settings or specificity of cancer cell types. Accordingly, one study demonstrated ICAM-1 inhibition to be associated with decreased invasion of breast cancer cells that exert a high baseline ICAM-1 expression (33). Other studies found anti-invasive properties of a-methylene-g-butyrolactone derivatives and thalidomide by suppressing ICAM-1 expression only in ICAM- 1-overexpressing A549 cells challenged with TNF (32, 34), which is not comparable to both cellular and stimulus conditions of our study. Only one of these studies revealed an antimetastatic effect of an ICAM-1 neutralizing antibody in A549 cells (34). However, these experiments are critical for comparison to the results of the present study, as they did not include comparison to a group treated with an appropriate isotype control antibody, the time frame of experiments was only 2 wk, and excessively high doses of ICAM-1 antibody (0.8 mg/g) were used, yielding a $3000-times higher dose when considering the body weight of nude mice between 20 to 30 g. Future research should address a potential ambivalent response of cells exhibiting different levels of basal ICAM-1 expression with respect to both proinvasive and anti-invasive stimuli. As ICAM-1 up-regulation in endothelial cells is supposed to promote vascular toxicity based on leukocyte/ endothelial interaction (55), a possible cannabinoidinduced ICAM-1 up-regulation in these cells may also serve as a risk factor. However, CB2 activation by HU-308 has been reported even to inhibit cisplatininduced leukocyte infiltration and ICAM-1 expression associated with damage of tubular structures in murine kidneys (56). Furthermore, CBD has been demonstrated to inhibit ICAM-1 expression in high-glucosetreated human coronary artery endothelial cells (57) without altering basal ICAM-1 expression, suggesting that the observed ICAM-1 induction may be restricted to cancer cells. In view of such tissue-specific regulations, it is, furthermore, tempting to speculate that the cannabinoid-induced TIMP-1 expression in cancer cells, and thereby at the microenviromental hotspots of cancer cell invasion, may serve as a more promising antimetastatic strategy as compared to direct matrix metalloproteinase (MMP) inhibitors. In fact, the MMP inhibitors marimastat and prinomastat revealed rather unsatisfactory clinical results, probably due to dose limitation because of musculoskeletal adverse reactions (58–60). Collectively, this study provides several important findings for the first time: a cannabinoid receptor and TRPV1-dependent ICAM-1 induction that exhibits a pivotal role in the TIMP-1-dependent anti-invasive action of cannabinoids; ICAM-1 as a molecular key player of an anti-invasive mechanism; anticancer properties of cannabinoids investigated in primary human lung tumor cells; and an inhibitor-based antimetastatic mechanism of cannabinoids in vivo. To assess the efficacy of cannabinoid-based drugs as systemic anticancer strategies, clinical trials are suggested.
This study was supported by a grant from the Deutsche Forschungsgemeinschaft (Hi 813/6-1).
REFERENCES
1. Jacobsson, S. O., Wallin, T., and Fowler, C. J. (2001) Inhibitionof rat C6 glioma cell proliferation by endogenous and syntheticcannabinoids. Relative involvement of cannabinoid and vanilloid receptors. J. Pharmacol. Exp. Ther. 299, 951–9592. Bla´zquez, C., Carracedo, A., Barrado, L., Real, P. J., Ferna´ndezLuna, J. L., Velasco, G., Malumbres, M., and Guzma´n, M. (2006)Cannabinoid receptors as novel targets for the treatment ofmelanoma. FASEB J. 20, 2633–26353. Bla´zquez, C., Casanova, M. L., Planas, A., Go´mez Del Pulgar, T.,Villanueva, C., Ferna´ndez-Acen˜ero, M. J., Aragone´s, J., Huffman, J. W., Jorcano, J. L., and Guzma´n, M. (2003) Inhibition oftumor angiogenesis by cannabinoids. FASEB J. 17, 529–5314. Bla´zquez, C., Gonza´lez-Feria, L., Alvarez, L., Haro, A., Casanova,M. L., and Guzma´n, M. (2004) Cannabinoids inhibit the vascular endothelial growth factor pathway in gliomas. Cancer Res. 64,5617–56235. Pisanti, S., Borselli, C., Oliviero, O., Laezza, C., Gazzerro, P., andBifulco, M. (2007) Antiangiogenic activity of the endocannabinoid anandamide: correlation to its tumor-suppressor efficacy.J. Cell. Physiol. 211, 495–5036. Galve-Roperh, I., Sa´nchez, C., Corte´s, M. L., del Pulgar, T. G.,Izquierdo, M., and Guzma´n, M. (2000) Antitumoral action ofcannabinoids: involvement of sustained ceramide accumulationand extracellular signal-regulated kinase activation. Nat. Med. 6,313–3197. Hinz, B., Ramer, R., Eichele, K., Weinzierl, U., and Brune, K.(2004) Up-regulation of cyclooxygenase-2 expression is involvedin R(1)-methanandamide-induced apoptotic death of humanneuroglioma cells. Mol. Pharmacol. 66, 1643–16518. Salazar, M., Carracedo, A., Salanueva, I. J., Herna´ndez-Tiedra,S., Lorente, M., Egia, A., Va´zquez, P., Bla´zquez, C., Torres, S.,García, S., Nowak, J., Fimia, G. M., Piacentini, M., Cecconi, F.,Pandolfi, P. P., Gonza´lez-Feria, L., Iovanna, J. L., Guzma´n, M.,Boya, P., and Velasco, G. (2009) Cannabinoid action inducesautophagy-mediated cell death through stimulation of ER stressin human glioma cells. J. Clin. Invest. 119, 1359–1372
9. Bla´zquez, C., Salazar, M., Carracedo, A., Lorente, M., Egia, A.,Gonza´lez-Feria, L., Haro, A., Velasco, G., and Guzma´n, M.(2008) Cannabinoids inhibit glioma cell invasion by downregulating matrix metalloproteinase-2 expression. Cancer Res.68, 1945–195210. Preet, A., Ganju, R. K., and Groopman, J. E. (2008) D9-Tetrahydrocannabinol inhibits epithelial growth factor-inducedlung cancer cell migration in vitro as well as its growth andmetastasis in vivo. Oncogene 27, 339–34611. Ramer, R., and Hinz, B. (2008) Inhibition of cancer cellinvasion by cannabinoids via increased expression of tissueinhibitor of matrix metalloproteinases-1. J. Natl. Cancer Inst. 100,59–6912. McAllister, S. D., Christian, R. T., Horowitz, M. P., Garcia, A.,and Desprez, P. Y. (2007) Cannabidiol as a novel inhibitor ofId-1 gene expression in aggressive breast cancer cells. Mol.Cancer Ther. 6, 2921–292713. Ramer, R., Merkord, J., Rohde, H., and Hinz, B. (2010) Cannabidiol inhibits cancer cell invasion via upregulation of tissueinhibitor of matrix metalloproteinases-1. Biochem. Pharmacol. 79,955–96614. Ramer, R., Rohde, A., Merkord, J., Rohde, H., and Hinz, B.(2010) Decrease of plasminogen activator inhibitor-1 may contribute to the anti-invasive action of cannabidiol on human lungcancer cells. Pharm. Res. 27, 2162–217415. Nithipatikom, K., Endsley, M. P., Isbell, M. A., Falck, J. R.,Iwamoto, Y., Hillard, C. J., and Campbell, W. P. (2004) 2-Arachidonoylglycerol: a novel inhibitor of androgen-independentprostate cancer cell invasion. Cancer Res. 64, 8826–883016. Xian, X. S., Park, H., Cho, Y. K., Lee, I. S., Kim, S. W., Choi,M. G., Chung, I. S., Han, K. H., and Park, J. M. (2010) Effect ofa synthetic cannabinoid agonist on the proliferation and invasion of gastric cancer cells. J. Cell. Biochem. 110, 321–33217. Rog, D. J., Nurmikko, T. J., and Young, C. A. (2007) OromucosalD9-tetrahydrocannabinol/cannabidiol for neuropathic pain associated with multiple sclerosis: an uncontrolled, open-label,2-year extension trial. Clin. Ther. 29, 2068–207918. Kogan, N. M., Bla´zquez, C., Alvarez, L., Gallily, R., Schlesinger,M., Guzma´n, M., and Mechoulam, R. (2006) Cannabinoidquinone inhibits angiogenesis by targeting vascular endothelialcells. Mol. Pharmacol. 70, 51–5919. Ligresti, A., Moriello, A. S., Starowicz, K., Matias, I., Pisanti, S.,De Petrocellis, L., Laezza, C., Portella, G., Bifulco, M., and DiMarzo, V. (2006) Antitumor activity of plant cannabinoids withemphasis on the effect of cannabidiol on human breast carcinoma. J. Pharmacol. Exp. Ther. 318, 1375–138720. McKallip, R. J., Jia, W., Schlomer, J., Warren, J. W., Nagarkatti,P. S., and Nagarkatti, M. (2006) Cannabidiol-induced apoptosisin human leukaemia cells: a novel role of cannabidiol in theregulation of p22phox and Nox4 expression. Mol. Pharmacol. 70,897–90821. Massi, P., Vaccani, A., Ceruti, S., Colombo, A., Abbracchio,M. P., and Parolaro, D. (2004) Antitumor effects of cannabidiol,a nonpsychoactive cannabinoid, on human glioma cell lines.J. Pharmacol. Exp. Ther. 308, 838–84522. Hubbard, A. K., and Rothlein, R. (2000) Intercellular adhesionmolecule-1 (ICAM-1) expression and cell signaling cascades.Free Radic. Biol. Med. 28, 1379–138623. Melis, M., Spatafora, M., Melodia, A., Pace, E., Gjomarkaj, M.,Merendino, A. M., and Bonsignore, G. (1996) ICAM-1 expression by lung cancer cell lines: effects of upregulation bycytokines on the interaction with LAK cells. Eur. Respir. J. 9,1831–183824. Sunami, T., Yashiro, M., and Chung, K. H. (2000) ICAM-1(intercellular adhesion molecule-1) gene transfection inhibitslymph node metastasis by human gastric cancer cells. Jpn. J.Cancer Res. 91, 925–93325. Kelly C. P., O’Keane J. C., Orellana J., Schroy P. C. 3rd, YangS., LaMont, J. T., and Brady, H. R. (1992) Human coloncancer cells express ICAM-1 in vivo and support LFA-1-dependent lymphocyte adhesion in vitro. Am. J. Physiol. 263,G864 –G87026. Vanky, F., Wang, P., Patarroyo, M., and Klein, E. (1990)Expression of the adhesion molecule ICAM-1 and major histocompatibility complex class I antigens on human tumor cells isrequired for their interaction with autologous lymphocytes invitro. Cancer Immunol. Immunother. 31, 19–2727. Altomonte, M., Gloghini, A., Bertola, G., Gasparollo, A., Carbone, A., Ferrone, S., and Maio, M. (1993) Differential expression of cell adhesion molecules CD54/CD11a and CD58/CD2by human melanoma cells and functional role in their interaction with cytotoxic cells. Cancer Res. 53, 3343–334828. Sawada, T., Kimura, K., Nishihara, T., Onoda, N., Teraoka, H.,Yamashita, Y., Yamada, N., Yashiro, M., Ohira, M., and Hirakawa, K. (2006) TGF-beta1 down-regulates ICAM-1 expressionand enhances liver metastasis of pancreatic cancer. Adv. Med.Sci. 51, 60–6529. Ogawa, Y., Hirakawa, K., Nakata, B., Fujihara, T., Sawada, T.,Kato, Y., Yoshikawa, K., and Sowa, M. (1998) Expression ofintercellular adhesion molecule-1 in invasive breast cancerreflects low growth potential, negative lymph node involvement,and good prognosis. Clin. Cancer Res. 4, 31–3630. Fujihara, T., Yashiro, M., Inoue, T., Sawada, T., Kato, Y., Ohira,M., Nishiguchi, Y., Ishikawa, T., Sowa, M., and Chung, K. H.(1999) Decrease in ICAM-1 expression on gastric cancer cells iscorrelated with lymph node metastasis. Gastric Cancer 2, 221–22531. Maeda, K., Kang, S. M., Sawada, T., Nishiguchi, Y., Yashiro, M.,Ogawa, Y., Ohira, M., Ishikawa, T., and Hirakawa-YS Chung, K.(2002) Expression of intercellular adhesion molecule-1 andprognosis in colorectal cancer. Oncol. Rep. 9, 511–51432. Huang, W. C., Chan, S. T., Yang, T. L., Tzeng, C. C., and Chen,C. C. (2004) Inhibition of ICAM- 1 gene expression, monocyteadhesion and cancer cell invasion by targeting IKK complex:Molecular and functional study of novel alpha-methylene-gamma-butyrolactone derivatives. Carcinogenesis 25, 1925–193433. Rosette, C., Roth, R. B., Oeth, P., Braun, A., Kammerer, S.,Ekblom, J., and Denissenko, M. F. (2005) Role of ICAM1 ininvasion of human breast cancer cells. Carcinogenesis 26, 943–95034. Lin, Y. C., Shun, C. T., Wu, M. S., and Chen, C. C. (2006) Anovel anticancer effect of thalidomide: inhibition of intercellular adhesion molecule-1-mediated cell invasion and metastasisthrough suppression of nuclear factor-kB. Clin. Cancer Res. 12,7165–717335. Miele, M. E., Bennett, C. F., Miller, B. E., and Welch, D. R.(1994) Enhanced metastatic ability of TNF-a-treated malignantmelanoma cells is reduced by intercellular adhesion molecule-1(ICAM-1, CD54) antisense oligonucleotides. Exp. Cell Res. 214,231–24136. Koyama, Y., Tanaka, Y., Saito, K., Abe, M., Nakatsuka, K.,Morimoto, I., Auron, P. E., and Eto, S. (1996) Cross-linking ofintercellular adhesion molecule 1 (CD54) induces AP-1 activation and IL-1b transcription. J. Immunol. 157, 5097–510337. Sano, H., Nakagawa, N., Chiba, R., Kurasawa, K., Saito, Y., andIwamoto, I. (1998) Cross-linking of intercellular adhesion molecule-1 induces interleukin-8 and RANTES production throughthe activation of MAP kinases in human vascular endothelialcells. Biochem. Biophys. Res. Commun. 250, 694–69838. Lawson, C., Ainsworth, M., Yacoub, M., and Rose, M. (1999)Ligation of ICAM-1 on endothelial cells leads to expression ofVCAM-1 via a nuclear factor-kB-independent mechanism. J. Immunol. 162, 2990–299639. Blaber, R., Stylianou, E., Clayton, A., and Steadman, R. (2003)Selective regulation of ICAM-1 and RANTES gene expressionafter ICAM-1 ligation on human renal fibroblasts. J. Am. Soc.Nephrol. 14, 116–12740. Edwards, D. R., Rocheleau, H., Sharma, R. R., Wills, A. J., Cowie,A., Hassell, J. A., and Heath, J. K. (1992) Involvement of AP1and PEA3 binding sites in the regulation of murine tissueinhibitor of metalloproteinases-1 (TIMP-1) transcription.Biochim. Biophys. Acta 1171, 41–5541. Bugno, M., Graeve, L., Gatsios, P., Koj, A., Heinrich, P. C.,Travis, J., and Kordula, T. (1995) Identification of the interleukin-6/oncostatin M response element in the rat tissue inhibitorof metalloproteinases-1 (TIMP-1) promoter. Nucleic Acids Res.23, 5041–504742. Ramer, R., Eichele, K., and Hinz, B. (2007) Upregulation oftissue inhibitor of matrix metalloproteinases-1 confers the antiinvasive action of cisplatin on human cancer cells. Oncogene 26,5822–582743. Hinz, B., Ro¨sch, S., Ramer, R., Tamm, E. R., and Brune, K.(2005) Latanoprost induces matrix metalloproteinase-1 expression in human nonpigmented ciliary epithelial cells through acyclooxygenase-2-dependent mechanism. FASEB J. 19, 1929–1931
44. Mukherjee, S., Adams, M., Whiteaker, K., Daza, A., Kage, K.,Cassar, S., Meyer, M., and Yao, B. B. (2004) Species comparisonand pharmacological characterization of rat and human CB2cannabinoid receptors. Eur. J. Pharmacol. 505, 1–945. Izzo, A. A., Borrelli, F., Capasso, R., Di Marzo, V., and Mechoulam, R. (2009) Non-psychotropic plant cannabinoids: new therapeutic opportunities from an ancient herb. Trends Pharmacol.Sci. 30, 515–52746. Consroe, P., Kennedy, K., and Schram, K. (1991) Assay ofplasma cannabidiol by capillary gas chromatography/ion trapmass spectroscopy following high-dose repeated daily oral administration in humans. Pharmacol. Biochem. Behav. 40, 517–52247. Scaldaferri, F., Sans, M., Vetrano, S., Correale, C., Arena, V.,Pagano, N., Rando, G., Romeo, F., Potenza, A. E., Repici, A.,Malesci, A., and Danese, S. (2009) The role of MAPK ingoverning lymphocyte adhesion to and migration across themicrovasculature in inflammatory bowel disease. Eur. J. Immunol. 39, 290–30048. Sapna, S., and Shivakumar, K. (2007) Hypoxia and antioxidantsenhance soluble ICAM-1 release from cardiac fibroblasts. Mol.Cell. Biochem. 303, 259–26249. Lee, H. M., Kim, H. J., Won, K. J., Choi, W. S., Lee, K. Y., Bae,Y. M., Park, P. J., Park, T. K., Lee, Y. L., Lee, C. K., and Kim, B.(2008) Contribution of soluble intercellular adhesion molecule-1 to the migration of vascular smooth muscle cells. Eur.J. Pharmacol. 579, 260–26850. Chen, C., Chou, C., Sun, Y., and Huang, W. (2001) Tumornecrosis factor alpha-induced activation of downstream NF-kBsite of the promoter mediates epithelial ICAM-1 expression andmonocyte adhesion. Involvement of PKCa, tyrosine kinase, andIKK2, but not MAPKs, pathway. Cell. Signal. 13, 543–55351. Radisavljevic, Z., Avraham, H., and Avraham, S. (2000) Vascularendothelial growth factor up-regulates ICAM-1 expression viathe phosphatidylinositol 3 OH-kinase/AKT/Nitric oxide pathway and modulates migration of brain microvascular endothelial cells. J. Biol. Chem. 275, 20770–2077452. Natali, P., Nicotra, M. R., Cavaliere, R., Bigotti, A., Romano, G.,Temponi, M., and Ferrone, S. (1990) Differential expression ofintercellular adhesion molecule 1 in primary and metastaticmelanoma lesions. Cancer Res. 50, 1271–127853. Hosch, S. B., Meyer, A. J., Schneider, C., Stoecklein, N., Prenzel,K. L., Pantel, K., Broelsch, C. E., and Izbicki, J. R. (1997)Expression and prognostic significance of HLA class I, ICAM-1, and tumor-infiltrating lymphocytes in esophageal cancer. J. Gastrointest. Surg. 1, 316–32354. Shin, H. S., Jung, C. H., Park, H. D., and Lee, S. S. (2004) Therelationship between the serum intercellular adhesion molecule-1 level and the prognosis of the disease in lung cancer.Korean J. Intern. Med. 19, 48–5255. Yu, M., Han, J., Cui, P., Dai, M., Li, H., Zhang, J., and Xiu, R.(2008) Cisplatin up-regulates ICAM-1 expression in endothelialcell via a NF-kB dependent pathway. Cancer Sci. 99, 391–39756. Mukhopadhyay, P., Rajesh, M., Pan, H., Patel, V., Mukhopadhyay, B., Ba´tkai, S., Gao, B., Hasko´ G, and Pacher, P. (2010)Cannabinoid-2 receptor limits inflammation, oxidative/nitrosative stress, and cell death in nephropathy. Free Radic. Biol. Med.48, 457–46757. Rajesh, M., Mukhopadhyay, P., Ba´tkai, S., Hasko´ G, Liaudet, L.,Drel, V. R., Obrosova, I. G., and Pacher, P. (2007) Cannabidiolattenuates high glucose-induced endothelial cell inflammatoryresponse and barrier disruption. Am. J. Physiol. Heart Circ. Physiol.293, H610–H61958. Shepherd, F. A., Giaccone, G., Seymour, L., Debruyne, C.,Bezjak, A., Hirsh, V., Smylie, M., Rubin, S., Martins, H., Lamont,A., Krzakowski, M., Sadura, A., and Zee, B. (2002) Prospective,randomized, double-blind, placebo-controlled trial of marimastat after response to first-line chemotherapy in patients withsmall-cell lung cancer: a trial of the National Cancer Institute ofCanada-Clinical Trials Group and the European Organizationfor Research and Treatment of Cancer. J. Clin. Oncol. 20,4434–443959. Bramhall, S. R., Schulz, J., Nemunaitis, J., Brown, P. D., Baillet,M., and Buckels, J. A. (2002) A double-blind placebo-controlled,randomised study comparing gemcitabine and marimastat withgemcitabine and placebo as first line therapy in patients withadvanced pancreatic cancer. Br. J. Cancer 87, 161–16760. Bissett, D., O’Byrne, K. J., von Pawel, J., Gatzemeier, U., Price,A., Nicolson, M., Mercier, R., Mazabel, E., Penning, C., Zhang,M. H., Collier, M. A., and Shepherd, F. A. (2005) Phase III studyof matrix metalloproteinase inhibitor prinomastat in non-smallcell lung cancer. J. Clin. Oncol. 23, 842–849Received for publication October 17, 2011. Accepted for publication December 5, 2011.