Prevention of Alzheimer’s Disease Pathology by Cannabinoids: Neuroprotection Mediated by Blockade of Microglial Activation
This article originally appeared at https://www.jneurosci.org/content/25/8/1904.abstract
Authors: Belén G. Ramírez, Cristina Blázquez, Teresa Gómez del Pulgar, Manuel Guzmán and María L. de Ceballos
Journal of Neuroscience 23 February 2005, 25 (8) 1904-1913; DOI: https://doi.org/10.1523/JNEUROSCI.4540-04.2005
Abstract
Alzheimer's disease (AD) is characterized by enhanced β-amyloid peptide (βA) deposition along with glial activation in senile plaques, selective neuronal loss, and cognitive deficits. Cannabinoids are neuroprotective agents against excitotoxicity in vitro and acute brain damage in vivo. This background prompted us to study the localization, expression, and function of cannabinoid receptors in AD and the possible protective role of cannabinoids after βA treatment, both in vivo and in vitro. Here, we show that senile plaques in AD patients express cannabinoid receptors CB1 and CB2, together with markers of microglial activation, and that CB1-positive neurons, present in high numbers in control cases, are greatly reduced in areas of microglial activation. In pharmacological experiments, we found that G-protein coupling and CB1 receptor protein expression are markedly decreased in AD brains. Additionally, in AD brains, protein nitration is increased, and, more specifically, CB1 and CB2 proteins show enhanced nitration. Intracerebroventricular administration of the synthetic cannabinoid WIN55,212-2 to rats prevent βA-induced microglial activation, cognitive impairment, and loss of neuronal markers. Cannabinoids (HU-210, WIN55,212-2, and JWH-133) block βA-induced activation of cultured microglial cells, as judged by mitochondrial activity, cell morphology, and tumor necrosis factor-α release; these effects are independent of the antioxidant action of cannabinoid compounds and are also exerted by a CB2-selective agonist. Moreover, cannabinoids abrogate microglia-mediated neurotoxicity after βA addition to rat cortical cocultures. Our results indicate that cannabinoid receptors are important in the pathology of AD and that cannabinoids succeed in preventing the neurodegenerative process occurring in the disease.
Introduction
Alzheimer's disease (AD), the most common form of dementia, is characterized by the deposition of β-amyloid peptide (βA) within one of its pathological hallmarks: the senile plaque. Activated microglia cluster at senile plaques (McGeer et al., 1987; Dickson et al., 1988), and this seems to be responsible for the ongoing inflammatory process in the disease. Transgenic mouse models of AD also develop plaques in which βA deposits and activated microglia exist (Masliah et al., 1996; Frautschy et al., 1998; Jantzen et al., 2002). Furthermore, microglial activation results in neurodegeneration both in vitro (Meda et al., 1995; Gao et al., 2002; Xie et al., 2002) and in vivo (Weldon et al., 1998; Herrera et al., 2000; Iravani et al., 2002) paradigms. In this context, recent studies have focused on the therapeutic interest of limiting microglial activation and inflammation in AD and other neurological disorders.
Cannabinoids, the active components of marijuana and their analogs, exert a wide spectrum of central and peripheral effects by activating specific cannabinoid receptors, two of which have been well characterized to date: CB1 and CB2 (Howlett et al., 2002; Piomelli, 2003). CB1 receptors are found in high density in the nervous system (Herkenham et al., 1990), in which they mediate cannabinoid psychoactivity, and all types of neural cells express them. Thus, in addition to being present in neurons, CB1 receptors exist in astrocytes (Bouaboula et al., 1995; Sánchez et al., 1998), microglia (Waksman et al., 1999; Walter et al., 2003), and oligodendrocytes (Molina-Holgado et al., 2002). In contrast, the CB2 receptor is considered to be expressed solely in cells and organs of the immune system and is unrelated to cannabinoid psychoactivity. There are also recent reports on the existence of CB2 receptors in microglia (Walter et al., 2003) and on cannabinoids affecting migration (Walter et al., 2003), as well as nitric oxide (NO) and cytokine production (Waksman et al., 1999; Puffenbarger et al., 2000; Facchinetti et al., 2003) in microglial cell cultures in vitro.
Cannabinoids exert neuroprotection under different experimental conditions. Thus, cannabinoid receptor activation protects hippocampal or granule cerebellar neurons from excitotoxicity (Skaper et al., 1996; Shen and Thayer, 1998; Hampson and Grimaldi, 2001) and from hypoxia and glucose deprivation (Nagayama et al., 1999). In vivo, cannabinoids decrease hippocampal neuronal loss and infarct volume after cerebral ischemia (Nagayama et al., 1999), acute brain trauma (Panikashvili et al., 2001), and ouabain-induced excitotoxicity (van der Stelt et al., 2001). These effects have been ascribed to inhibition of glutamate transmission, reduction of calcium influx, and subsequent inhibition of noxious cascades, such as tumor necrosis factor-α (TNF-α) generation and oxidative stress.
This background prompted us to study the characteristics and localization of cannabinoid receptors in AD brain, with particular emphasis on any relationship with microglial activation. Furthermore, the effects of cannabinoid receptor activation were studied in an animal model of AD in vivo and in a model of βA-induced microglial activation in vitro.
Materials and Methods
Materials. βA25-35 and a peptide containing the same 11 amino acids but with a scrambled (SCR) sequence (NeoMPS, Strasbourg, France) were used throughout. βA25-35 and the scrambled peptide were dissolved in distilled water at a concentration of 2.5 mg/ml to allow self-aggregation of the peptide and stored at -80°C until used. βA1-40 (NeoMPS) was dissolved in PBS (1.72 mg/ml), aged at 37°C for 24 h (“fibrillar” peptide), and was vortexed several times during that period, and aliquots were stored at -80°C until used. The control (C) peptide was not subjected to aging (“soluble” peptide). Aggregation of all of the peptides was confirmed by microscopy after staining with Coomassie brilliant blue (Fernández-Tomé et al., 2004). WIN55,212-2 [R-(+)-(2,3-dihydro-5-methyl-3-[(4-morpholinyl)methyl]1,2,3-de]-1,4-benzoxazin-6-yl)(1-naphthalenyl) methanone sulfonate] was obtained from Sigma (St. Louis, MO), JWH-133 [(6aR,10aR),-3-(1,1-dimethylbutyl)-6a,7,10,10a-tetrahydro-6,6,9-trimethyl-6H-dibenzo[b,d]pyran] was a generous gift from Dr. J. W. Huffman (Clemson University, Clemson, SC), HU-210 [(6aR,10aR)-3-(1,1′-dimethylheptyl)-6a,7,10,10a-tetrahydro-1-hydroxy-6,6-dimethyl-6H-dibenzo[b,d]pyran-9-methanol] was kindly given by Dr. R. Mechoulam (The Hebrew University of Jerusalem, Jerusalem, Israel), AM251 [N-1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-N-1-piperidinyl-1H-pyrazole-3-carboxamide] was from Tocris Cookson (Bristol, UK), and SR141716 [N-piperidino-5-(4-chloro-phenyl)-1-(2,4-dichlorophenyl)-4-methyl-3-pyrazole carboxamide] and SR144528 [N-[(1S)-endo-1,3,3-trimethyl bicyclo[2.2.1]heptan-2-yl]-5-(4-chloro-3-methylphenyl)-1-(4-methylbenzyl)-pyrazole-3-carbo-xamide] were kindly donated by Sanofi-Synthelabo (Montpellier, France). Each of these compounds was dissolved in DMSO at 10 mm concentration, and aliquots were stored at -80°C, with the exception of WIN55,212-2, which was initially dissolved in chloroform (on ice), quickly aliquoted to prevent evaporation, and dried under a stream of N2, and aliquots were stored desiccated. Before their use, drugs were diluted in the appropriate solvent (e.g., PBS or cell culture medium), and DMSO never exceeded 0.1% in pharmacological or cell culture experiments. Cell culture reagents were from Sigma unless otherwise stated. Salts and other reactives were analytical grade from Merck (Darmstadt, Germany).
Patient samples. For immunocytochemistry, cryoprotected and fixed frozen samples from frontal cortex were obtained from the Neurologic Tissue Bank (Hospital Clinic, Barcelona, Spain). Controls consisted of three males and two females (mean ± SEM; 62.8 ± 7.2 years of age; 9.5 ± 2.2 h of postmortem interval), and clinically diagnosed and neuropathologically defined AD patients consisted of three females and three males (76.0 ± 3.3 years of age; 12.5 ± 3.1 h of postmortem interval). For biochemical studies, frozen frontal cortex samples (control, n = 18; AD, n = 18) were obtained from the London Brain Bank for Neurodegenerative Diseases (Institute of Psychiatry, London, UK). Control subjects and AD patients were matched for age (C, 71.8 ± 2.6; AD, 77.4 ± 2.2 years), sex (13 females and 5 males per group), and postmortem delay (C, 38.7 ± 4.7; AD, 33.3 ± 5.3 h). Samples were maintained at -80°C until assayed.
Immunocytochemistry. Immunostaining was performed on floating sections (30 μm) as described previously (Gómez del Pulgar et al., 2002). For CB2 and N-Tyr immunostaining, antigen retrieval was achieved by boiling sections in 1 mm EDTA, pH 8.0, for 5 min before the standard labeling technique. Sections were incubated with the different antibodies overnight at 4°C. Dilutions of antibodies were as follows: polyclonal anti-CB1 (1:900; from Dr. K. Mackie, University of Washington, Seattle, WA), polyclonal anti-CB2 (1:900; Affinity BioReagents, Golden, CO), polyclonal anti-N-Tyr [1:000; Dr. J. Rodrigo, Cajal Institute, Consejo Superior de Investigaciones Científicas (CSIC), Madrid, Spain] (Uttenthal et al., 1998), monoclonal anti-GFAP (1:1000; Sigma), monoclonal anti-human leukocyte antigen-D region related (HLA-DR) (1:15; MP Biomedicals, Irvine, CA), and biotinylated tomato lectin (TL) (1:150; Sigma). Development was conducted by the ABC method (Pierce, Rockford, IL), and immunoreactivity was visualized by 3,3′-diaminobenzidine oxidation as chromogen, with or without nickel enhancement.
Immunostaining of cell cultures, either neuronal or microglial, were similarly performed after fixation with paraformaldehyde (4% in 0.1 m phosphate buffer) for 30 min, followed by rinses with PBS. In addition, rat microglia was immunostained with monoclonal anti-rat CD11 (OX42; 1:100; Serotec, Oxford, UK).
Omission of primary or secondary antibodies resulted in no immunostaining. Specificity of anti-CB1 and anti-CB2 staining was performed by preabsorption of the antibodies with the antigenic peptides (kindly given by Dr. K. Mackie), which completely abolished labeling.
3H-WIN55,212-2 and 35S-GTPγS binding. 3H-WIN55,212-2 binding was conducted as described previously (Breivogel et al., 2001). Briefly, P2 membrane fractions (80 μg of protein) were incubated in assay buffer (Tris 50 mm, pH 7.4, containing 3 mm MgCl2, 1 mm EDTA, and 1 mg/ml fatty acid-free BSA) with 3 nm 3H-WIN55,212-2 (specific activity, 41.0 Ci/mmol; PerkinElmer Life Sciences, Boston, MA) at 30°C for 60 min. Nonspecific binding was determined in the presence of 1 μm SR141716. Saturation curves were constructed with increasing concentrations of 3H-WIN55,212-2 (0.25-10 nm), and, in competition curves, several concentrations of the agonists or antagonists (10-10 to 10-4 m) were assayed. The binding was terminated by filtering through GF/C Whatman (Maidstone, UK) filters (0.45 μm), incubated previously with 0.5% polyethyleneimine for at least 2 h, using a Brandel (Gaithersburg, MD) harvester, washed three times with ice-cold 5 mm Tris HCl, pH 7.4, containing 1 mg/ml BSA, and the radioactivity was counted using an LKB-Wallac (Gaithersburg, MD) scintillation counter. 35S-GTPγS binding was performed as described previously (Breivogel et al., 2001). P2 membrane fractions (30 μg of protein) were incubated in assay buffer (50 mm Tris HCl, pH 7.4, containing 3 mm MgCl2, 100 mm Na Cl, 0.2 mm EGTA, and 0.5 mg/ml fatty acid-free BSA) with 0.04 nm 35S-GTPγS (specific activity, 1250 Ci/mmol; PerkinElmer Life Sciences), 30 μm GDP, and 5 μm WIN55,212-2 at 30°C for 60 min. Binding was terminated by rapid filtration as above. Basal activity was determined in the absence of agonist and nonspecific binding in the presence of 10 μm of unlabeled GTPγS. Net stimulation was the difference of the binding in the presence and in the absence (basal binding) of WIN55,212-2. Data were expressed in femtomoles per milligram of protein. All of the procedures for both assays were conducted in plasticware and dilutions of radioligands and cold reagents in plastic tubes coated with 50 mm Tris HCl, pH 7.4, containing 1 mg/ml BSA.
βA and cannabinoid administration to rats. All of the experiments were performed according to ethical regulations on the use and welfare of experimental animals of the European Union and the Spanish Ministry of Agriculture, and the procedures were approved by the bioethical committee of the CSIC. βA administration to male Wistar rats was performed essentially as described previously (Pavía et al., 2000). βA25-35 or a peptide with a scrambled sequence (NeoMPS), which was used as control, were injected intracerebroventricularly daily for 7 d (20 μg in 10 μl of saline per day), and the animals were tested at different times after the first injection. Other animals received a cannabinoid (WIN55,212-2, 10 μg in 10 μl of 20% DMSO/80% saline per day) together with the peptides. The Hamilton syringe used for intracerebroventricular injections was repeatedly washed with distilled water, followed by flushing with 1 mg/ml BSA solution, which reduces drastically binding to glass. This procedure was performed before every injection.
Behavioral tests. All of the behavioral procedures were conducted at the same time of the day (9:00 A.M. to 2:00 P.M.). Motor activity was monitored in four activity cages (Digiscan; AccuScan Instruments, Columbus, OH) in an isolated room for 30 min, immediately after injections at days 1 or 7 of treatment, or at 2 months after treatment initiation. Horizontal motor activity and number of stereotypies and rearings were recorded. To determine spatial learning, rats were trained to find a hidden platform in a water tank of 150 cm of diameter. Four trials per day with different start positions, each 30 min apart, were conducted for 5 d (Müller et al., 1994), and latency to reach the platform was recorded. Cutoff time to find the platform was 120 s, and rats failing to find the platform were placed on it and left there for 15 s. Data acquisition was performed with a video camera (Noldus Information Technology, Wageningen, The Netherlands).
Cell cultures and treatments. Primary mixed glial cultures were prepared from neonatal rat cortex as described previously (McCarthy and de Vellis, 1980). Mechanically dissociated cortices were seeded onto 75 cm2 flasks in DMEM/Ham's F-12, supplemented with 10% fetal calf serum (FCS) and 40 μg/ml gentamicine. Cells were cultured in a humified atmosphere of 5% CO2/95% air at 37°C, and the medium was changed every 2 or 3 d. After being cultured for 3 weeks, flasks were shaken for 2-3 h at 230 rpm, and floating cells were pelleted, seeded onto a plastic dish, and incubated at 37°C for 4 h, and loosely bound cells were aspirated. Adherent cells were detached (PBS plus 1 mm EDTA) and 3 × 104 cells were seeded onto poly-Orn-coated 96-well plates in DMEM/Ham's F-12 supplemented with 0.5% FCS. The cultures were at least 99% pure, as judged by immunocytochemical criteria. Mitochondrial activity (redox state) was assessed by the MTT assay. Neuron cultures from rat cortices were prepared as described previously (Fernández-Tomé et al., 2004) with some modifications. Briefly, cerebral cortices were mechanically dissociated in trypsin (1 mg/ml; Worthington, Freehold, NJ) at 37°C for 10 min, followed by DNaseI addition (50 μl; Roche Products, Welwyn Garden City, UK). Cells were collected after centrifugation and seeded further in Neurobasal (Invitrogen, San Diego, CA), containing B27 additives (Invitrogen), onto gelatin/poly-Lys-coated P24 plates (Falcon, Franklin Lakes, NJ) at a density of 25 × 104 cells. After 4 d in culture, they were switched to MEM with N2 additives (Sigma), and treatments (6 d in vitro) were performed in MEM. Neuron cultures were completely devoid of microglia as determined by TL or OX42 immunostaining (data not shown). For cocultures, 15 × 104 microglial cells were seeded in inserts (membrane pore size, 0.4 μm; diameter, 9 mm; Costar, Cambridge, MA) in DMEM containing 0.5% FCS and, at 24 h, were treated with βA1-40 for 4 h to avoid direct toxicity of the peptide on neurons, and the medium was aspirated and placed over neuron cultures (25 × 104; 24-well plates, Costar) for an additional 20 h. Drugs were added in  of the final volume to maintain aggregation of peptides. Cell counts were performed by Coomassie brilliant blue (0.2% in 10% acetic acid/40% methanol) staining under phase-contrast microscopy by an observer unaware of the treatments (four fields per condition in triplicate) in a Zeiss (Oberkochen, Germany) Axiovert microscope. Neurons showing intact neurites with uniform diameter and soma with a smooth round appearance were considered viable, whereas neurons with fragmented neurites and a shrunken cell body were considered nonviable.
of the final volume to maintain aggregation of peptides. Cell counts were performed by Coomassie brilliant blue (0.2% in 10% acetic acid/40% methanol) staining under phase-contrast microscopy by an observer unaware of the treatments (four fields per condition in triplicate) in a Zeiss (Oberkochen, Germany) Axiovert microscope. Neurons showing intact neurites with uniform diameter and soma with a smooth round appearance were considered viable, whereas neurons with fragmented neurites and a shrunken cell body were considered nonviable.
TNF-α analysis. Cell-free supernatants from microglial cultures were collected, microfuged, stored at -80°C, and assayed by a TNF-α commercial sandwich ELISA (Biosource, Camarillo, CA) in strict accordance with instructions of the manufacturer. The sensitivity was 10 pg/ml.
Western blot analysis. Western blot was performed as described previously (Molina-Holgado et al., 2002). Tissues were sonicated in lysis buffer, samples were centrifuged at high speed for 10 min, and supernatants were collected. Total protein was assessed by the Bio-Rad (Hercules, CA) protein assay. An aliquot of each sample (30 μg of protein) was separated by SDS-PAGE (10%), and proteins were transferred from the gels onto nitrocellulose membranes. The blots were blocked with 1% defatted dry milk for 1 h at room temperature and incubated overnight at 4°C with the following antibodies: anti-CB1 (1:5000), anti-CB2 (1:2000), anti-N-Tyr (1:3000), polyclonal anti-calbindin D-28K (1:5000; Swant, Bellinzona, Switzerland), and monoclonal anti-α-tubulin (1:20,000; Sigma). Finally, samples were subjected to enhanced chemiluminescence and densitometric analysis. Densitometric analysis of bands was performed by Quantity One quantitation software (version 4.2; Bio-Rad) from film exposures; the background was always subtracted, and the percentage of optical density was obtained considering 100% of that of control samples within the same film. Immunoprecipitations were performed by incubating overnight at 4°C lysate aliquots (100 μg of protein) with the anti-N-Tyr antibody (1 μg/mg protein lysate) prebound to protein A-agarose (Sigma). After washing, the immunoprecipitates were resolved by 10% SDS-PAGE and Western blotted with anti-CB1 and anti-CB2 antibodies as above.
Statistical analysis. Statistical significance analysis was assessed by using two-way or one-way ANOVA, followed by Bonferroni's post hoc test or by unpaired Student's t test (Prism software, version 4.0; GraphPad Software, San Diego, CA). A value of p < 0.05 was considered significant. Binding saturation and inhibition curves were plotted by nonlinear regression, and IC50 values were determined using the one-site competition model (Prism software).
Results
Cannabinoid receptors in AD brain
We studied by immunolabeling the localization of cannabinoid receptors in relation to senile plaques and microglial activation. All of the AD cases studied (six of six) showed CB1 and CB2 immunoreactivity in frontal cortical senile plaques (Fig. 1a), along with markers of microglial activation, such as the histocompatibility glycoprotein HLA-DR (McGeer et al., 1987) (Fig. 1a) and protein nitration (Fig. 1a, N-Tyr). Double-immunolabeling studies addressed the question of whether CB1 neurons are vulnerable in AD, and microglial activation accounted for it. In agreement with previous immunohistochemical studies in nonhuman primates (Ong and Mackie, 1999), high numbers of CB1 neurons and fibers, surrounded by resting microglia, were observed in all layers of the frontal cortex in controls (Fig. 1b). CB1-positive neuron density was greatly reduced in AD. Thus, although in some AD cases (two of six) CB1-positive neurons were still present in areas of microglial activation (Fig. 1b), they were completely absent in others (four of six) (Fig. 1b). Likewise, no coexistence of CB1-positive plaques and neurons was evident in these cases. In addition to CB2 expression in AD plaques, we also found labeling in tangle-like neurons and dystrophic neurites (four of six cases) (Fig. 1c), whereas normal brain was devoid of any signal (five of five cases). Labeling specificity was demonstrated by antigenic peptide preabsorption of the anti-CB antibodies used (Fig. 1c), which completely blocked immunoreactivity.

{kind=link}
{kind=link}
Figure 1.
Cannabinoid receptor localization in AD brain. a, CB1 and CB2 immunostaining in senile plaques, along with the markers of microglial activation HLA-DR and N-Tyr. b, Double immunostaining of HLA-DR (black, arrows) and CB1 (brown, asterisks). CB1-positive neurons in controls (top); CB1-positive neurons are still present (middle) or completely lost (bottom) in areas of intense microglial activation in AD. c, CB1-positive and CB2-positive neurons and dystrophic neurites in AD. Insets, Absence of labeling by preabsorption of the antibodies with the antigenic peptide (pep). Scale bars, 25 μm.
AD brain protein nitration (Smith et al., 1996) is thought to be a consequence of the reaction of NO and superoxide to form the toxic peroxynitrite radical. In control brain, N-Tyr immunoreactivity was present in astrocytes and in neuronal nuclei (Fig. 2a). N-Tyr-positive astrocytes were present in AD brain as well (data not shown), but now many pyramidal neurons showed cytoplasmic labeling in all of the cases studied (six of six) (Fig. 2a). Protein nitration was significantly increased in AD compared with control brain (Fig. 2b). Moreover, nitration of CB1 and CB2 protein was markedly increased in AD, as shown by immunoprecipitation studies (Fig. 2c).

{kind=link}
{kind=link}
Figure 2.
Nitration of CB1 and CB2 is increased in AD brain. a, N-Tyr-immunoreactive astrocytes in control (top); nuclear N-Tyr expression in control (middle); cytoplasmic N-Tyr expression in AD (arrows, bottom). Scale bar, 25 μm. b, Total protein nitration (as detected by Western blot) in control (C) and AD brain. OD, Optical density. c, Lysates from control and AD brains were immunoprecipitated with anti-N-Tyr antibody and blotted with anti-CB1 or CB2 antibodies. The percentage of nitration of total CBs is shown. b, c, Results are mean ± SEM of n = 18 in each group; **p < 0.01 and ***p < 0.001 compared with controls (Student's t test); representative blots are shown. Error bars represent SEM.
Subsequently, we examined the pharmacological characteristics of cannabinoid receptors by using the synthetic cannabinoid agonist WIN55,212-2. 3H-WIN55,212-2-specific binding, as defined by the selective CB1 antagonist SR141716 (Rinaldi-Carmona et al., 1994) at 1 μm, was similar in control and AD cases (Fig. 3a), as were the saturation curves for the binding process (Fig. 3b). Indeed, the density of receptors (Bmax; control, 318.1 ± 87.5 fmol/mg protein; AD, 304.7 ± 62.2 fmol/mg protein; n = 3) and the apparent affinity of the binding (Kd; control, 3.72 ± 0.83 nm; AD, 5.36 ± 0.62 nm; n = 3) were unaltered in AD. Indeed, there was considerable overlap of data points among controls and AD patients. Although basal 35S-GTPγS binding was unchanged in AD cortex (Fig. 3c), WIN55,212-2-stimulated 35S-GTPγS binding, which measures G-protein coupling and therefore cannabinoid receptor activation, was dramatically decreased (-63%) in AD (Fig. 3d). There was no correlation between age, sex, or postmortem parameters and the values of binding, in neither controls nor AD patients. Analysis of cannabinoid receptor levels by Western blot showed that CB1 protein expression was reduced in AD (Fig. 3e), whereas no changes in CB2 expression were observed (Fig. 3f).

{kind=link}
{kind=link}
Figure 3.
CB1 receptor binding is unaltered and G-protein coupling is reduced in AD frontal cortex. a, Specific 3H-WIN55,212-2 (WIN) binding (n = 18 in each group). b, Representative 3H-WIN55212-2 binding saturation curves (n = 3 in each group). c, Basal 35S-GTPγS binding (n = 18 in each group). d, WIN55,212-2-stimulated 35S-GTPγS binding (n = 18; *p < 0.05 compared with controls; Student's t test). e, f, CB1 (e) and CB2 (f) expression (as detected by Western blot) in control and AD brain. OD, Optical density. Results are mean ± SEM of n = 18 in each group; *p < 0.05 compared with controls (Student's t test); representative blots are shown. Error bars represent SEM.
WIN55,212-2 is a mixed CB1/CB2 cannabinoid receptor agonist that activates, with similar affinities, CB1 and CB2 receptors (Howlett et al., 2002) and may also interact with other, as yet uncharacterized, receptor subtypes (Breivogel et al., 2001). Therefore, its pharmacological selectivity was assessed in normal human brain. 3H-WIN55,212-2 binding was completely displaced by the CB1-selective antagonists SR141716 and AM251 (Lan et al., 1999) (pEC50, 6.88 ± 0.14 and 6.22 ± 0.12, respectively) but was not affected by the selective CB2 antagonist SR144528 (Rinaldi-Carmona et al., 1998) and the CB2-selective agonist JWH-133 (Huffman et al., 1999). 35S-GTPγS binding was stimulated by WIN55,212-2 (maximal effect over basal, 87.9 ± 20.2%; pEC50, 6.31 ± 0.05), not affected by JWH-133 (up to 5 μm), and completely blocked by SR141716 and AM251 (pEC50, 7.87 ± 0.13 and 7.87 ± 0.10, respectively). Together, these results indicate that, in human brain, WIN55,212-2 interacts with CB1 or CB1-like receptors.
Cannabinoid treatment prevents βA-induced toxic effects in vivo
βA overexpression or administration to rodents models AD by activating microglia (Masliah et al., 1996; Frautschy et al., 1998; Netland et al., 1998) and inducing cognitive impairment (Müller et al., 1994; Delobette et al., 1997). We administered βA (20 μg/d, i.c.v) or the control peptide (SCR) alone or in combination with WIN55,212-2 (10 μg/d) and studied microglia staining at the end of the treatment (8 d). Resting microglia were observed in rats injected with SCR and/or the cannabinoid (Fig. 4). Interestingly, there was an intense microglial activation in the cortex of βA-treated rats, which was prevented by WIN55,212-2 treatment (Fig. 4).

{kind=link}
{kind=link}
Figure 4.
Cannabinoid treatment prevents βA-induced microglial activation in rats. Tomato lectin binding to microglial cells in frontal cortex of rats 24 h after treatment completion was increased by βA compared with SCR peptide and prevented by WIN55,212-2 (WIN) cotreatment; pictures of one representative animal of three per group are shown. Initial magnification was 200×.
We next examined whether cannabinoid administration affected cognitive function. In contrast to control rats that learned the spatial navigation task over 5 d of training, βA-treated rats failed to do so. More important, WIN55,212-2 treatment prevented βA-induced cognitive impairment (Fig. 5a), whereas it did not alter the learning process when combined with SCR. This cannabinoid effect was not attributable to changes in the locomotor activity of the animals. Thus, WIN55,212-2 produced its typical hypolocomotor action on the first day of treatment, but this effect was not evident at the seventh day of administration and 2 months later (data not shown), when the navigation task was performed. As expected, intracerebroventricular administration of the cannabinoid agonist did not induce adverse effects. Indeed, just after the cessation of the treatment, the general hematological profiles of the WIN55,212-2-treated rats were normal. Likewise, neither the biochemical parameters nor markers for tissue damage changed at the end of the 7 d administration period.

{kind=link}
{kind=link}
Figure 5.
Cannabinoid treatment prevents cognitive impairment and loss of neuronal markers in rats. a, Latency (in seconds) to find a hidden platform in the water maze during training. Results are mean of n = 5 in each group; SEM have been omitted for clarity and were always <12% of the mean; *p < 0.05 and **p < 0.01 compared with SCR-treated rats at the same training day; #p < 0.05 and ##p < 0.01 compared with βA-treated rats (ANOVA with Bonferroni's post hoc test). WIN, WIN55,212-2. b, c, Expression of calbindin (b) and α-tubulin (c) in control (C) and AD frontal cortex; results are mean ± SEM of n = 18 control and AD; ***p < 0.001 compared with controls. d-f, Expression of CB1 (d), calbindin (e), and α-tubulin (f) in frontal cortex of rats at 2 months after treatment. OD, Optical density. Results are mean ± SEM of n = 5 in each group; *p < 0.05 and **p < 0.01 compared with SCR-treated rats (ANOVA with Bonferroni's post hoc test); representative blots are shown. Error bars represent SEM.
Interestingly, the changes in neuronal protein expression observed in AD patients were mimicked by repeated administration of βA to rats. Thus, the expression of the neuronal markers calbindin (Fig. 5b,e) and α-tubulin (Fig. 5c,f) was significantly reduced in both situations. Of importance, this reduction was attenuated by cannabinoid administration to rats (Fig. 5e,f). CB1 expression was also decreased in βA-treated rats (Fig. 5d), as was the case in AD brain (Fig. 3e).
Cannabinoid treatment prevents βA-induced microglial activation and neurotoxicity in vitro
First, we examined the effects of cannabinoids on βA-induced alterations in pure microglial cell cultures. As expected, microglia in culture expressed both CB1 and CB2 receptors (Walter et al., 2003) (Fig. 6a). Microglial activation after fibrillar βA challenge (0.5 μm) included morphological changes at 24 h (Fig. 6b), as well as increased mitochondrial activity (in the absence of cell proliferation; data not shown) and TNF-α release (Meda et al., 1995; Casal et al., 2002) at 4 h (Fig. 6c) and 24 h (data not shown). The cannabinoid agonist HU-210 (100 nm), which alone had no significant effect on these parameters, counteracted fibrillar βA-induced microglial activation. Indeed, the rod-like morphology with lamellipodia acquired by fibrillar βA-treated cells (Casal et al., 2002) turned to the resting oval morphology by HU-210 cotreatment (Fig. 6b). HU-210 also prevented the enhancement in TNF-α release observed after fibrillar βA addition. This effect was mimicked by WIN55,212-2 (100 nm), a cannabinoid devoid of antioxidant properties (Marsicano et al., 2002), and JWH-133 (100 nm), a CB2-selective agonist (Huffman et al., 1999) devoid of psychoactive effects when administered in vivo (Sánchez et al., 2001). In summary, cannabinoids counteract βA-mediated activation of microglia in culture.

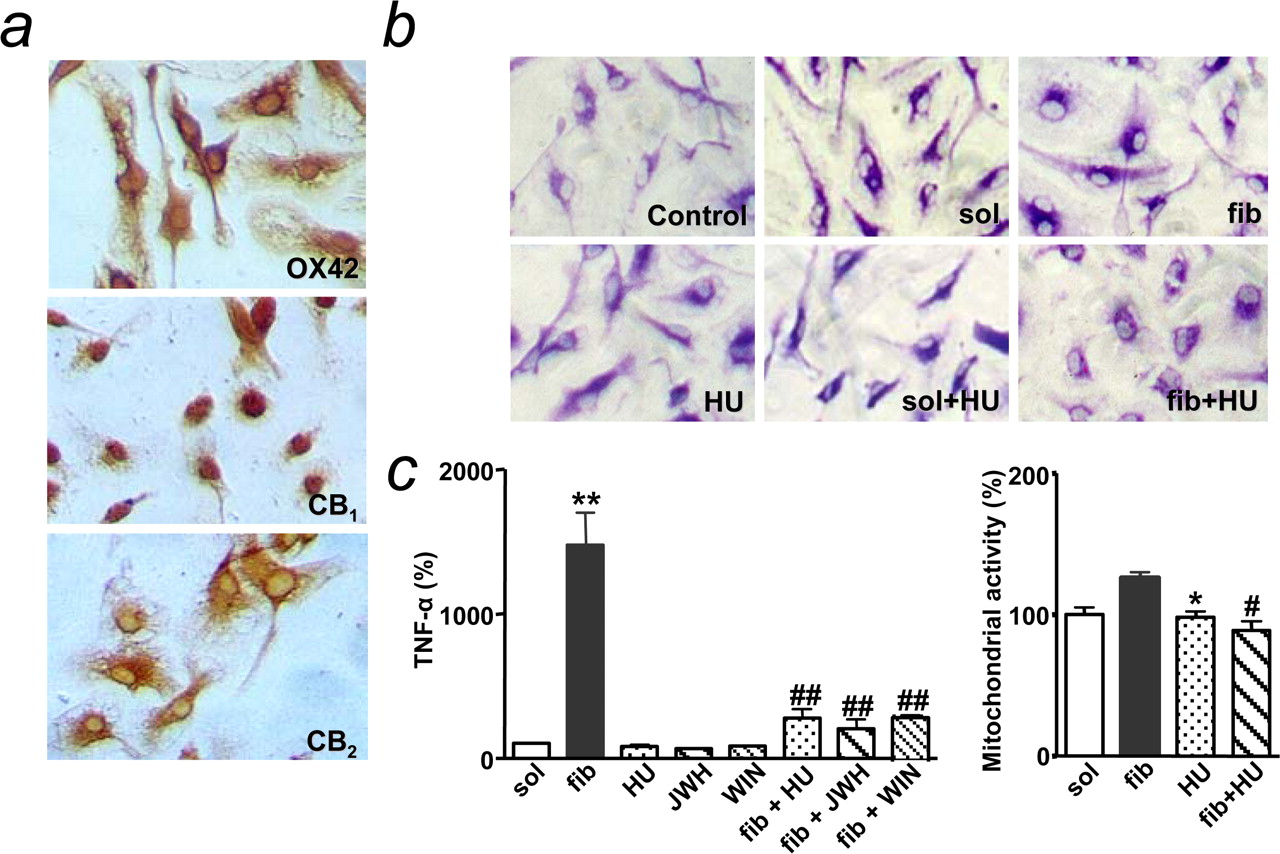{kind=link}
{kind=link}
Figure 6.
Cannabinoids prevent βA-induced microglial activation in vitro. a, Immunostaining of cultured microglia with anti-OX42 (top), CB1 (middle), and CB2 (bottom) antibodies. b, Fibrillar βA1-40 (fib), but not soluble βA1-40 (sol), induced a rod-like morphology, which was prevented by HU-210 (HU). c, Cannabinoids [HU-210 (HU), WIN55,212-2 (WIN), and JWH-133 (JWH), at 100 nm for 4 h] prevented TNF-α release and mitochondrial activity, as induced by fibrillar βA1-40 (500 nm). TNF-α release in controls was 26.1 ± 4.5 pg/ml. Results are mean ± SEM of n = 4-6; *p < 0.05 and **p < 0.01 compared with soluble βA1-40-treated control cultures; #p < 0.05, ##p < 0.01 compared with fibrillar βA1-40-treated cultures (ANOVA with Bonferroni's post hoc test).
βA exerts direct toxicity on neurons, but indirect neurotoxicity through microglia-mediated activation has been observed as well (Meda et al., 1995; Tan et al., 2000; Xie et al., 2002). Cannabinoids were unable to prevent direct toxicity of high concentrations (1-10 μm) of βA in primary cortical neurons in culture (data not shown). Subsequently, we assessed indirect βA neurotoxicity through microglia activation in cocultures. To avoid any direct toxicity to neurons, microglia seeded in inserts was treated with the peptides or the cannabinoids for 4 h, a time sufficient for microglia activation. Indeed, there was significant neurotoxicity when neurons were exposed for 20 h to microglial cells pretreated with 0.5 μm fibrillar βA for 4 h (Fig. 7a,b), whereas, at this time point and concentration, the peptide did not significantly alter neuron survival in the absence of microglial cells (Fig. 7a) (Barger et al., 1995; Fernández-Tomé et al., 2004). Both WIN55,212-2 and JWH-133 (100 nm) prevented microglia-mediated neurotoxicity after βA treatment (Fig. 7a,b). The neuroprotective effect of WIN55,212-2 on βA neurotoxicity in cocultures was prevented by the selective antagonists SR141716 and SR144528, whereas the effect of JWH-133 was only counteracted by the latter (Fig. 7a). These results support that the neuroprotective effect of cannabinoids relies on the prevention of βA-induced microglial activation.

{kind=link}
{kind=link}
Figure 7.
Cannabinoids prevent βA-induced microglia-mediated neurotoxicity in vitro. a, Neurons (neu) were treated with fibrillar βA1-40 (fib) or soluble βA1-40 (sol) alone (500 nm) or in combination with cannabinoid agonists [WIN55,212-2 (WIN) or JWH-133 (JWH), 100nm] and antagonists [SR141716 (SR1) or SR144528 (SR2), 100 nm] for 20 h (top); alternatively, microglia (mg) seeded in inserts were treated with the same compounds for 4 h, media were removed, and inserts were placed on neurons for 20 h (middle and bottom). Neurons were fixed, stained with Coomassie brilliant blue, and counted. Results are mean ± SEM of n = 3. Middle, *p < 0.05 compared with soluble βA1-40-treated control cultures; #p < 0.05 compared with fibrillar βA1-40-treated cultures (ANOVA with Bonferroni's post hoc test). Bottom, The set of three columns on the left represents single treatments (fibrillar βA1-40, SR141716, or SR144528); *p < 0.05 compared with cultures treated with fibrillar βA1-40 alone; #p < 0.05 compared with fibrillar βA1-40 plus the cannabinoid agonist. b, Microglia-neuron cocultures showing neurotoxicity after exposure to fibrillar βA1-40-activated microglia and prevention by cannabinoids.
Discussion
One of the key features of AD is microglial activation, a process that may serve dual functions. For a long time, it was thought to be responsible for the ongoing inflammatory process occurring in the neurological condition (for review, see Akiyama et al., 2000). However, it has been recognized recently that removal of βA by activated microglia may be beneficial (Bard et al., 2000; Wyss-Coray et al., 2001; Jantzen et al., 2002). These cells are part of the senile plaques in cerebral cortex and hippocampus of the afflicted patients. We have shown that CB1 and CB2 receptors are colocalized in plaques, in line with a recent report (Benito et al., 2003). Upregulation of inducible nitric oxide synthase occurs in senile plaques in AD (Lee et al., 1999) and in microglia after βA administration in vivo (Weldon et al., 1998). Furthermore, N-Tyr immunoreactivity has been observed in plaques in AD (Vodovotz et al., 1996) and in mice carrying amyloid precursor protein plus presenilin-1 mutations (Matsuoka et al., 2001). Protein nitration therefore appears to be a marker of the effects of the peroxynitrite radical and a consequence of microglial activation. In normal human brain, we observed a high density of CB1-positive neurons (Ong and Mackie, 1999; Benito et al., 2003), mostly pyramidal cells, which are lost in AD brain, in particular in areas bearing plaques, pointing to the vulnerability of such neurons to the toxic species generated by microglia (e.g., NO and cytokines). Given that CB-positive neurons were segregated from plaques, their possible contribution to CB receptors in plaques may be ruled out, and this points to microglial cells as the unique source.
Previous works in human brain have mapped CB receptors by autoradiographic techniques (Westlake et al., 1994; Glass et al., 1997, 2000), but their pharmacological characteristics were not determined. Furthermore, it is widely accepted that CB2 receptors are not present in normal brain, but we wondered whether, by using an appropriate selection of cold ligands to define specific binding, we would be able to show CB2 receptors, at least in pathological brain, such as AD. In human cortical membranes, 3H-WIN55,212-2 binding to CB1 receptors, as defined by the CB1-selective antagonist SR141716, was saturable and of high affinity. The apparent affinity is similar to that observed in rat brain (Breivogel et al., 1997), albeit the density of binding sites (Bmax) is much lower. Rather than a species trait, aging (Mailleux and Vanderhaeghen, 1992; Romero et al., 1998; Mato and Pazos, 2004) may account for the difference in density, given that our controls were nonpathological aged subjects. WIN55,212-2 is a mixed CB1/CB2 cannabinoid receptor agonist that activates, with similar affinities, CB1 and CB2 receptors (Howlett et al., 2002) and may also interact with other, as yet uncharacterized, receptor subtypes (Breivogel et al., 2001). The relative potencies of the drugs in our pharmacological studies are in general agreement with previous studies (Rinaldi-Carmona et al., 1994, 1998; Breivogel et al., 1997), including those in cells transfected with the cloned human cannabinoid receptors (Felder et al., 1995; MacLennan et al., 1998). Together, these results indicate that, in human brain, WIN55,212-2 interacts with CB1 or CB1-like receptors. CB1 receptors outnumber other characterized cannabinoid receptors, and such a high density may have masked CB2 receptors, which were observed by immunohistochemistry, with a very restricted localization in AD brain.
In a previous work, cannabinoid receptor binding in cortical areas of AD brain was unchanged, and the reduction in hippocampal subfields was unrelated to the pathological changes (Westlake et al., 1994). However, in agreement with our histological findings, in biochemical studies, we have shown that CB1 protein expression and function are markedly reduced in AD. Indeed G-protein coupling, as judged by 35S-GTPγS binding, was greatly diminished in samples from AD patients. Given that enhanced protein nitration was observed along CB receptors in plaques, we examined whether they may be a target for such alteration. In AD, total protein nitration is increased compared with controls (Aoyama et al., 2000). More specifically, both CB1 and CB2 receptor proteins show enhanced protein nitration in AD than in controls. It should be noted that protein nitration, known to inactivate other proteins in AD (Aoyama et al., 2000), might interfere with CB receptor function as well. In summary, significant alterations in the localization, expression, and function of cannabinoid receptors occur in AD and may play a role in its physiopathology.
Our present results confirm and extend those of previous works, showing marked alterations in cannabinoid receptors in AD brain. Benito et al. (2003) reported that CB1-positive neurons appeared to be preserved in AD parahippocampal cortex, but frontal cortex was not studied. Therefore, the region of study may account for the difference between their and our results. Autoradiographic studies, used by Westlake et al. (1994), lack cellular resolution, which may explain why decreased binding did not correlate with areas showing pathology (senile plaques and tangles) in hippocampal subfields in AD. In this work, we combined histological, pharmacological, and biochemical techniques to show loss of CB1-positive neurons in frontal cortex of AD brain, as well as decreased CB1 protein expression and G-protein coupling, despite preserved binding.
In the present study, we used rats repeatedly injected intracerebroventricularly with βA to model AD. Indeed, we reported decreased muscarinic receptors in cortex (Pavía et al., 2000) and increased monomeric G1 acetylcholinesterase isoform (Sáez-Valero et al., 2002), similar to those in AD. These animals show increased microglial reaction at the end of βA administration (Netland et al., 1998; Weldon et al., 1998) and cognitive impairment at 2 months (Delobette et al., 1997) compared with control animals. Interestingly, βA-treated rats reproduced the same reduction in neuronal markers, including CB1 protein, occurring in AD, validating further the experimental model. Cannabinoids induce neuroprotection against excitotoxicity (Shen and Thayer, 1998), ischemia (Nagayama et al., 1999), and glucose deprivation (Nagayama et al., 1999) in vitro. These compounds are also effective against acute brain damage in vivo, including excitotoxic insult (van der Stelt et al., 2001), ischemia (Nagayama et al., 1999), and acute brain trauma (Panikashvili et al., 2001). Furthermore, the involvement of CB1 receptors and their endogenous ligands as neuroprotectants has been demonstrated recently in CB1-null mutants, which are more susceptible to neurodegeneration (Parmentier-Batteur et al., 2002; Marsicano et al., 2003). In this work, we report that microglial activation induced in vivo by βA was completely prevented by cannabinoid administration. We have shown that the loss of neuronal markers induced by βA is attenuated by cannabinoid administration. More importantly, the cognitive deficits occurring in βA-treated rats are also prevented by the cannabinoid. Together, these results constitute the first evidence that cannabinoids exert neuroprotection in a model of AD.
Cannabinoids prevent microglial activation and decrease NO production (Waksman et al., 1999) and TNF-α expression and release (Puffenbarger et al., 2000; Facchinetti et al., 2003) evoked by different agents. In this work, cannabinoids with different pharmacological profiles effectively counteracted the well known βA-induced microglial activation, in particular TNF-α release, which is potentially neurotoxic. Milton (2002) reported that the toxicity of high concentrations (25 μm) of βA peptides was prevented by endocannabinoids in NT-2 cells, a human teratocarcinoma cell line that can be differentiated into neuronal phenotype, and in myeloma cells. Thus, anandamide and noladin ether protected the cells by a CB1- or a CB1/CB2-mediated effect, respectively, depending on the cell line used. These results were not reproduced in our neuronal cultures. Thus, different synthetic cannabinoids did not prevent direct toxicity of βA on primary neurons in culture. However, a neuroprotective effect of cannabinoids was disclosed using a βA microglia-mediated neurotoxicity paradigm. The selective CB2 agonist JWH-133 was as effective as WIN55,212-2, the mixed CB1/CB2 agonist. As shown in microglia-neuron cocultures and in βA-treated rats, these beneficial effects of cannabinoids may rely on their ability to block βA-induced microglial activation. In line with this notion, cannabinoids are unable to prevent βA-induced death of primary neurons in culture. This may be of particular importance for the possible endorsement of cannabinoids to therapeutic applications because of their psychotropic side effects, which may cast clinical concern. CB2 receptors appear to be exclusively expressed by microglia, and blockade of its activation may be attained by CB2 agonists with no overt psychoactivity.
The search for new and effective treatments for AD is of crucial importance, and limiting ongoing inflammatory responses secondary to microglial activation has been proposed (Akiyama et al., 2000). Furthermore, recent studies on therapeutic strategies for neurodegenerative diseases such as Parkinson's disease and AD have focused on the neuroprotective properties (e.g., slowing the ongoing neurodegeneration) rather than just on palliating symptoms of the diseases (Dawson and Dawson, 2002). Because cannabinoids combine both anti-inflammatory and neuroprotective actions, our findings may set the basis for the use of these compounds as a therapeutic approach for AD.
Footnotes
- This work was supported by Grants SAF 2002-01566 (M.L.C) and SAF 2003-00745 (M.G.) from the Spanish Ministry of Science and Technology, Grant CAM 08.1/0079 from the Community of Madrid (M.G., M.L.C.), Red de Investigación de Enfermedades Neurológicas (M.L.C.), and Fundación Científica de la Asociación Española contra el Cáncer (M.G.). B.G.R. is the recipient of a fellowship from the Community of Madrid. We thank Dr. K. Mackie for anti-CB1 antibody and CB1 and CB2 antigenic peptides, Dr. J. Rodrigo for anti-N-Tyr antibody, and Sanofi-Synthelabo for SR141716 and SR144528. Dr. I. Ferrer is acknowledged for helpful discussions, Dr. L. López-Mascaraque for assistance in microscopic imaging, and M. E. Fernández de Molina for excellent technical assistance.
- Correspondence should be addressed to Dr. María L. de Ceballos, Neurodegeneration Group, Cajal Institute, Consejo Superior de Investigaciones Científicas, Avenida Doctor Arce, 37, 28002 Madrid, Spain. E-mail: mceballos@cajal.csic.es.
- Copyright © 2005 Society for Neuroscience 0270-6474/05/251904-10$15.00/0
References
- ↵ Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy SA, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, et al. (2000) Inflammation and Alzheimer's disease. Neurobiol Aging 21: 383-421.CrossRefPubMedGoogle Scholar
- ↵ Aoyama K, Matsubara K, Fujikawa Y, Nagahiro Y, Shimizu K, Umegae N, Hayase N, Shiono H, Kobayashi S (2000) Nitration of manganese superoxide dismutase in cerebrospinal fluids is a marker for peroxynitrite-mediated oxidative stress in neurodegenerative diseases. Ann Neurol 47: 524-527.CrossRefPubMedGoogle Scholar
- ↵ Bard F, Cannon C, Barbour R, Burke R-L, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberbug I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, et al. (2000) Peripherally administered antibodies against amyloid β-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med 6: 916-919.CrossRefPubMedGoogle Scholar
- ↵ Barger SW, Hörster D, Furukawa K, Goodman Y, Krieglstein J, Mattson MP (1995) Tumor necrosis factors α and β protect neurons against amyloid β-peptide toxicity: evidence for involvement of κ B-binding factor and attenuation of peroxide and Ca2+ accumulation. Proc Natl Acad Sci USA 92: 9328-9332.Abstract/FREE Full TextGoogle Scholar
- ↵ Benito C, Núñez E, Tolón RM, Carrier EJ, Rabano A, Hillard CJ, Romero J (2003) Cannabinoid CB2 receptors and fatty acid amide hydrolase are selectively overexpressed in neuritic plaque-associated glia in Alzheimer's disease brains. J Neurosci 23: 11136-11141.Abstract/FREE Full TextGoogle Scholar
- ↵ Bouaboula M, Bourrie B, Rinaldi-Carmona M, Shire D, Le Fur G, Casellas P (1995) Stimulation of cannabinoid receptor CB1 induces krox-24 expression in human astrocytoma cells. J Biol Chem 270: 13973-13980.Abstract/FREE Full TextGoogle Scholar
- ↵ Breivogel CS, Sim LJ, Childers SR (1997) Regional differences in cannabinoid receptor/G-protein coupling in rat brain. J Pharmacol Exp Ther 282: 1632-1642.Abstract/FREE Full TextGoogle Scholar
- ↵ Breivogel CS, Griffin G, Di Marzo V, Martin BR (2001) Evidence for a new G protein-coupled cannabinoid receptor in mouse brain. Mol Pharmacol 60: 155-163.Abstract/FREE Full TextGoogle Scholar
- ↵ Casal C, Serratosa J, Tusell JM (2002) Relationship between β-AP peptide aggregation and microglial activation. Brain Res 928: 76-84.CrossRefPubMedGoogle Scholar
- ↵ Dawson TM, Dawson VL (2002) Neuroprotective and neurorestorative strategies for Parkinson's disease. Nat Neurosci 5: 1058-1061.Google Scholar
- ↵ Delobette S, Privat A, Maurice T (1997) In vitro aggregation facilitates β-amyloid peptide-(25-35)-induced amnesia in the rat. Eur J Pharmacol 319: 1-4.CrossRefPubMedGoogle Scholar
- ↵ Dickson DW, Farlo J, Davies P, Crystal H, Fuld P, Yen SH (1988) Alzheimer's disease. A double-labeling immunohistochemical study of senile plaques. Am J Pathol 132: 86-101.PubMedGoogle Scholar
- ↵ Facchinetti F, Del Giudice E, Furegato S, Passarotto M, Leon A (2003) Cannabinoids ablate release of TNFα in rat microglial cells stimulated with lypopolysaccharide. Glia 41: 161-168.CrossRefPubMedGoogle Scholar
- ↵ Felder CC, Joyce KE, Briley EM, Mansouri J, Mackie K, Blond O, Lai Y, Ma AL, Mitchell RL (1995) Comparison of the pharmacology and signal transduction of the human cannabinoid CB1 and CB2 receptors. Mol Pharmacol 48: 443-450.AbstractGoogle Scholar
- ↵ Fernández-Tomé P, Brera B, Arévalo M-A, de Ceballos ML (2004) β-Amyloid25-35 inhibits glutamate uptake in cultured neurons and astrocytes: modulation of uptake as a survival mechanism. Neurobiol Dis 15: 580-589.PubMedGoogle Scholar
- ↵ Frautschy SA, Yang F, Irrizarry M, Hyman B, Saido TC, Hsiao K, Cole GM (1998) Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathol 152: 307-317.PubMedGoogle Scholar
- ↵ Gao H-M, Hong J-S, Zhang W, Liu B (2002) Distinct role for microglia in rotenone-induced degeneration of dopaminergic neurons. J Neurosci 22: 782-790.Abstract/FREE Full TextGoogle Scholar
- ↵ Glass M, Dragunow M, Faull RLM (1997) Cannabinoid receptors in the human brain: a detailed anatomical and quantitative autoradiographic study in the fetal, neonatal and adult human brain. Neuroscience 77: 299-318.CrossRefPubMedGoogle Scholar
- ↵ Glass M, Dragunow M, Faull RLM (2000) The pattern of neurodegeneration in Huntington's disease: a comparative study of cannabinoid, dopamine, adenosine, and GABAA receptor alterations in the human basal ganglia in Huntington's disease. Neuroscience 97: 505-519.CrossRefPubMedGoogle Scholar
- ↵ Gómez del Pulgar T, de Ceballos ML, Guzmán M, Velasco G (2002) Cannabinoids protect astrocytes from ceramide-induced apoptosis through phosphatidylinositol 3-kinase/protein kinase B. J Biol Chem 277: 36527-36533.Abstract/FREE Full TextGoogle Scholar
- ↵ Hampson AJ, Grimaldi M (2001) Cannabinoid receptor activation and elevated cyclic AMP reduce glutamate neurotoxicity. Eur J Neurosci 13: 1529-1536.CrossRefPubMedGoogle Scholar
- ↵ Herkenham M, Lynn AB, Little DM, Johnson MR, Melvin LS, De Costa BR, Rice KC (1990) Cannabinoid receptor localization in brain. Proc Natl Acad Sci USA 87: 1932-1936.Abstract/FREE Full TextGoogle Scholar
- ↵ Herrera AJ, Castaño A, Venero JL, Cano J, Machado A (2000) The single intranigral injection of LPS as a new model for studying the selective effects of inflammatory reactions on dopaminergic systems. Neurobiol Dis 7: 429-447.CrossRefPubMedGoogle Scholar
- ↵ Howlett AC, Barth F, Bonner TI, Cabral G, Casellas P, Devane W, Herkenham M, Mackie K, Martin BR, Mechoulan R, Pertwee RG (2002) International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev 54: 161-202.Google Scholar
- ↵ Huffman JW, Liddle J, Yu S, Aung MM, Abood ME, Wiley JL, Martin BR (1999) 3-(1′,1′-Dimethylbutyl)-1-deoxy-delta8-THC and related compounds: synthesis and selective ligands for the CB2 receptor. Bioorg Med Chem 7: 2905-2914.CrossRefPubMedGoogle Scholar
- ↵ Iravani MM, Kashefi K, Mander P, Rose S, Jenner P (2002) Involvement of inducible nitric oxide synthase in inflammation-induced dopaminergic neurodegeneration. Neuroscience 110: 49-58.CrossRefPubMedGoogle Scholar
- ↵ Jantzen PT, Connor KE, DiCarlo G, Wenk GL, Wallace JL, Rojiani AM, Coppola D, Morgan D, Gordon MN (2002) Microglial activation and β-amyloid deposit reduction caused by nitric oxide-releasing nonsteroidal anti-inflammatory drug in amyloid precursor protein plus presenilin-1 transgenic mice. J Neurosci 22: 2246-2254.Abstract/FREE Full TextGoogle Scholar
- ↵ Lan R, Liu Q, Fan P, Lin S, Fernando SR, McCallion D, Pertwee R, Makriyannis A (1999) Structure-activity relationships of pyrazole derivatives as cannabinoid receptor antagonists. J Med Chem 42: 769-774.CrossRefPubMedGoogle Scholar
- ↵ Lee SC, Zhao M-L, Hirano A, Dickson DW (1999) Inducible nitric oxide synthase immunoreactivity in the Alzheimer disease hippocampus: association with Hirano bodies, neurofibrillary tangles and senile plaques. J Neuropathol Exp Neurol 58: 1163-1169.PubMedGoogle Scholar
- ↵ MacLennan SJ, Reynen PH, Kwan J, Bonhaus DW (1998) Evidence for inverse agonism of SR141716A at human recombinant cannabinoid CB1 and CB2 receptors. Br J Pharmacol 124: 619-622.CrossRefPubMedGoogle Scholar
- ↵ Mailleux P, Vanderhaeghen JJ (1992) Age-related loss of cannabinoid receptor binding sites and mRNA in the rat striatum. Neurosci Lett 147: 179-181.CrossRefPubMedGoogle Scholar
- ↵ Marsicano G, Moosmann B, Hermann HM, Lutz B, Behl C (2002) Neuroprotective properties of cannabinoids against oxidative stress: role of the cannabinoid receptor CB1. J Neurochem 80: 448-456.CrossRefPubMedGoogle Scholar
- ↵ Marsicano G, Goodenough S, Monory K, Hermann H, Eder M, Cannich A, Azad SC, Cascio MG, Gutierrez SO, van der Stelt M, López-Rodríguez ML, Casanova E, Schütz G, Zieglgänsberger W, Di Marzo V, Behl C, Lutz B (2003) CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science 302: 84-88.Abstract/FREE Full TextGoogle Scholar
- ↵ Masliah E, Sisk A, Mallory M, Mucke L, Schenk D, Games D (1996) Comparison of neurodegenerative pathology in transgenic mice overexpressing V717F β-amyloid precursor protein and Alzheimer's disease. J Neurosci 16: 5795-5811.Abstract/FREE Full TextGoogle Scholar
- ↵ Mato S, Pazos A (2004) Influence of age, postmortem delay and freezing storage period on cannabinoid receptor density and functionality in human brain. Neuropharmacology 46: 716-726.CrossRefPubMedGoogle Scholar
- ↵ Matsuoka Y, Picciano M, La Francois, Duff K (2001) Fibrillar β-amyloid evokes oxidative damage in a transgenic mouse model of Alzheimer's disease. Neuroscience 104: 609-613.CrossRefPubMedGoogle Scholar
- ↵ McCarthy KD, de Vellis J (1980) Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol 263: 17205-17208.Google Scholar
- ↵ McGeer PL, Itagaki S, Tago H, McGeer EG (1987) Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci Lett 79: 195-200.CrossRefPubMedGoogle Scholar
- ↵ Meda L, Cassatella MA, Szendrei GI, Otvos Jr L, Baron P, Villalba M, Ferrari D, Rossi F (1995) Activation of microglial cells by β-amyloid protein and interferon-γ. Nature 374: 647-650.CrossRefPubMedGoogle Scholar
- ↵ Milton NG (2002) Anandamide and noladin ether prevent neurotoxicity of the human amyloid-β peptide. Neurosci Lett 332: 127-130.CrossRefPubMedGoogle Scholar
- ↵ Molina-Holgado E, Vela JM, Arévalo-Martín A, Almazán G, Molina-Holgado F, Borrell J, Guaza C (2002) Cannabinoids promote oligodendrocyte progenitor survival: involvement of cannabinoid receptors and phosphatylinositol-3 kinase/Akt signaling. J Neurosci 22: 9742-9753.Abstract/FREE Full TextGoogle Scholar
- ↵ Müller U, Cristina N, Li Z-W, Wolfer DP, Lipp H-P, Rulicke T, Brandner S, Aguzzi T, Weissmann C (1994) Behavioral and anatomical deficits in mice homozygous for a modified β-amyloid precursor protein. Cell 79: 755-765.CrossRefPubMedGoogle Scholar
- ↵ Nagayama T, Sinor AD, Simon RP, Chen J, Graham SH, Jin K, Greenberg DA (1999) Cannabinoids and neuroprotection in global and focal cerebral ischemia and in neuronal cultures. J Neurosci 19: 2987-2995.Abstract/FREE Full TextGoogle Scholar
- ↵ Netland EE, Newton JL, Majocha RE, Tate BA (1998) Indomethacin reverses the microglial response to amyloid β-protein. Neurobiol Aging 19: 201-204.CrossRefPubMedGoogle Scholar
- ↵ Ong WY, Mackie K (1999) A light and electron microscopic study of the CB1 cannabinoid receptor in primate brain. Neuroscience 92: 1177-1191.CrossRefPubMedGoogle Scholar
- ↵ Panikashvili D, Simeonidou C, Ben-Shabat S, Hanus L, Breuer A, Mechoulam R, Shohami E (2001) An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature 413: 527-531.CrossRefPubMedGoogle Scholar
- ↵ Parmentier-Batteur S, Jin K, Mao XO, Xie L, Greenberg DA (2002) Increased severity of stroke in CB1 cannabinoid receptor knock-out mice. J Neurosci 22: 9771-9775.Abstract/FREE Full TextGoogle Scholar
- ↵ Pavía J, Alberch J, Alvarez I, Toledano A, de Ceballos ML (2000) Repeated intracerebroventricular administration of β-amyloid(25-35) to rats decreases muscarinic receptors in cerebral cortex. Neurosci Lett 278: 69-72.CrossRefPubMedGoogle Scholar
- ↵ Piomelli D (2003) The molecular logic of endocannabinoid signalling. Nat Rev Neurosci 4: 873-884.CrossRefPubMedGoogle Scholar
- ↵ Puffenbarger RA, Boothe AC, Cabral GA (2000) Cannabinoids inhibit LPS-inducible cytokine mRNA expression in rat microglial cells. Glia 29: 58-69.CrossRefPubMedGoogle Scholar
- ↵ Rinaldi-Carmona M, Barth F, Héaulme M, Shire D, Calandra B, Congy C, Martinez S, Maruani J, Néliat G, Caput D, Ferrara P, Soubrié P, Brelière JC, Le Fur G (1994) SR141716A, a potent and selective antagonist of the brain cannabinoid receptor. FEBS Lett 350: 240-244.CrossRefPubMedGoogle Scholar
- ↵ Rinaldi-Carmona M, Barth F, Millan J, Derocq JM, Casellas P, Congy C, Oustric D, Sarran M, Bouaboula M, Calandra B, Portier M, Shire D, Breliére JC, Le Fur GL (1998) SR144528, the first potent and selective antagonist of the CB2 cannabinoid receptor. J Pharmacol Exp Ther 284: 644-650.Abstract/FREE Full TextGoogle Scholar
- ↵ Romero J, Berrendero F, Garcia-Gil L, de la Cruz P, Ramos JA, Fernandez-Ruiz JJ (1998) Loss of cannabinoid receptor binding and messenger RNA and cannabinoid agonist-stimulated [35S]guanylyl-5′O-(thio)-triphosphate binding in the basal ganglia of aged rats. Neuroscience 84: 1075-1083.CrossRefPubMedGoogle Scholar
- ↵ Sáez-Valero J, de Ceballos ML, Small DH, de Felipe C (2002) Changes in molecular isoform distribution of acetylcholinesterase in rat cortex and cerebrospinal fluid after intracerebroventricular administration of amyloid β-peptide. Neurosci Lett 325: 199-202.CrossRefPubMedGoogle Scholar
- ↵ Sánchez C, Galve-Roperh I, Canova C, Brachet P, Guzmán M (1998) Δ9-tetrahydrocannabinol induces apoptosis in C6 glioma cells. FEBS Lett 436: 6-10.CrossRefPubMedGoogle Scholar
- ↵ Sánchez C, de Ceballos ML, Gómez del Pulgar T, Rueda D, Corbacho C, Velasco G, Galve-Roperh I, Huffman JW, Ramón y Cajal S, Guzmán M (2001) Inhibition of glioma growth in vivo by selective activation of the CB2 cannabinoid receptor. Cancer Res 61: 5784-5789.Abstract/FREE Full TextGoogle Scholar
- ↵ Shen M, Thayer SA (1998) Cannabinoid receptor agonists protect cultured rat hippocampal neurons from excitotoxicity. Mol Pharmacol 54: 459-462.Abstract/FREE Full TextGoogle Scholar
- ↵ Skaper SD, Buriani A, Dal Toso R, Petrelli L, Romanello S, Facci L, Leon A (1996) The ALIAmide palmitoylethanolamide and cannabinoids, but not anadamide, are protective in a delayed postglutamate paradigm of excitotoxic death in cerebellar granule neurons. Proc Natl Acad USA 93: 3984-3989.Abstract/FREE Full TextGoogle Scholar
- ↵ Smith MA, Harris PLR, Sayre LM, Beckman JS, Perry G (1996) Widespread peroxynitrite-mediated damage in Alzheimer's disease. J Neurosci 17: 2653-2657.Google Scholar
- ↵ Tan J, Town T, Mori T, Saxe Y, Crawford F, Mullan M (2000) CD45 opposes β-amyloid peptide-induced microglial activation via inhibition of p44/42 mitogen-activated protein kinase. J Neurosci 20: 7587-7594.Abstract/FREE Full TextGoogle Scholar
- ↵ Uttenthal LO, Alonso D, Fernández AP, Campbell RO, Moro MA, Leza JC, Lizasoain I, Esteban FJ, Barroso JB, Valderrama R, Pedrosa JA, Peinado MA, Serrano J, Richart A, Bentura ML, Santacana M, Martínez-Murillo R, Rodrigo J (1998) Neuronal and inducible nitric oxide synthase and nitrotyrosine immunoreactivities in the cerebral cortex of the aging rat. Microsc Res Tech 43: 75-88.CrossRefPubMedGoogle Scholar
- ↵ van der Stelt M, Veldhuis WB, Bär PR, Veldink GA, Vliegenthart JFG, Nicolay K (2001) Neuroprotection by Δ9-tetrahydrocannabinol, the main active compound of marijuana, against ouabain-induced in vivo excitotoxicity. J Neurosci 21: 6475-6479.Abstract/FREE Full TextGoogle Scholar
- ↵ Vodovotz Y, Lucia MS, Flanders KC, Chesler L, Xie, Q-W, Smith TW, Weidner J, Mumfors R, Webber R, Nathan C, Roberts AB, Lippa CF, Sporn MB (1996) Inducible nitric oxide synthase in tangle-bearing neurons of patients with Alzheimer's disease. J Exp Med 184: 1425-1433.Abstract/FREE Full TextGoogle Scholar
- ↵ Waksman Y, Olson J, Carlisle SJ, Cabral GA (1999) The central cannabinoid receptor (CB1) mediates inhibition of nitric oxide production by rat microglial cells. J Pharmacol Exp Ther 288: 1357-1366.Abstract/FREE Full TextGoogle Scholar
- ↵ Walter L, Franklin A, Witting A, Wade C, Xie Y, Kunos, G. Mackie K, Stella N (2003) Nonpsychotropic cannabinoid receptors regulate microglial cell migration. J Neurosci 23: 1398-1405.Abstract/FREE Full TextGoogle Scholar
- ↵ Weldon DT, Rogers SD, Ghilardi JR, Finke MP, Cleary JP, O'Hare E, Esler WP, Maggio JE, Mantyh PW (1998) Fibrillar β-amyloid induces microglial phagocytosis, expression of inducible nitric oxide synthase, and loss of a select population of neurons in the rat CNS in vivo. J Neurosci 18: 2161-2173.Abstract/FREE Full TextGoogle Scholar
- ↵ Westlake TM, Howlett AC, Bonner TI, Matsuda LA, Herkenham M (1994) Cannabinoid receptor binding and messenger RNA expression in human brain: an in vitro receptor autoradiography and in situ hybridization histochemistry study of normal aged and Alzheimer's brains. Neuroscience 63: 637-652.CrossRefPubMedGoogle Scholar
- ↵ Wyss-Coray T, Lin C, Yan F, Yu G-Q, Rohde B, McConlogue L, Masliah E, Mucke L (2001) TGF-β1 promotes microglial amyloid-β clearance and reduces plaque burden in transgenic mice. Nat Med 7: 612-618.CrossRefPubMedGoogle Scholar
- ↵ Xie Z, Wei M, Morgan TE, Fabrizio P, Han D, Finch CE, Longo VD (2002) Peroxynitrite mediates neurotoxicity of amyloid β-peptide1-42- and lipopolysaccharide-activated microglia. J Neurosci 22: 3484-3492.Abstract/FREE Full TextGoogle Scholar