
The endogenous cannabinoid, anandamide, induces cell death in colorectal carcinoma cells
This article originally appeared at https://gut.bmj.com/content/54/12/1741.abstract
Authors:
- H A Patsos1,
- D J Hicks1,
- R R H Dobson1,
- A Greenhough1,
- N Woodman1,
- J D Lane2,
- A C Williams1,
- C Paraskeva1
- Correspondence to:
Professor C Paraskeva
Cancer Research UK Colorectal Tumour Biology Group, Department of Pathology and Microbiology, School of Medical Sciences, University Walk, University of Bristol, Bristol BS8 1TD, UK
Abstract
Background and aims: Cyclooxygenase 2 (COX-2) is upregulated in most colorectal cancers and is responsible for metabolism of the endogenous cannabinoid, anandamide, into prostaglandin-ethanolamides (PG-EAs). The aims of this study were to determine whether anandamide and PG-EAs induce cell death in colorectal carcinoma (CRC) cells, and whether high levels of COX-2 in CRC cells could be utilised for their specific targeting for cell death by anandamide.
Methods: We determined the effect of anandamide on human CRC cell growth by measuring cell growth and cell death, whether this was dependent on COX-2 protein expression or enzyme activity, and the potential involvement of PG-EAs in induction of cell death.
Results: Anandamide inhibited the growth of CRC cell lines HT29 and HCA7/C29 (moderate and high COX-2 expressors, respectively) but had little effect on the very low COX-2 expressing CRC cell line, SW480. Induction of cell death in HT29 and HCA7/C29 cell lines was partially rescued by the COX-2 selective inhibitor NS398. Cell death induced by anandamide was neither apoptosis nor necrosis. Furthermore, inhibition of fatty acid amide hydrolase potentiated the non-apoptotic cell death, indicating that anandamide induced cell death was mediated via metabolism of anandamide by COX-2, rather than its degradation into arachidonic acid and ethanolamine. Interestingly, both PGE2-EA and PGD2-EA induced classical apoptosis.
Conclusions: These findings suggest anandamide may be a useful chemopreventive/therapeutic agent for colorectal cancer as it targets cells that are high expressors of COX-2, and may also be used in the eradication of tumour cells that have become resistant to apoptosis.
http://dx.doi.org/10.1136/gut.2005.073403
Statistics from Altmetric.com
Referenced in 1 Wikipedia pages
58 readers on Mendeley
Request Permissions
If you wish to reuse any or all of this article please use the link below which will take you to the Copyright Clearance Center’s RightsLink service. You will be able to get a quick price and instant permission to reuse the content in many different ways.
Colorectal cancer is one of the major causes of cancer death in the industrialised world, and the incidence is likely to rise even further with the increasing trend towards obesity. Therefore, it is essential to develop more prevention strategies and novel agents for colorectal cancer chemotherapy. Recently, there has been a great deal of interest in cannabinoids as novel anticancer agents.1 Cannabinoids, both plant derived (from Cannabis sativa) and endogenous, are compounds that have the ability to activate cannabinoid receptors: CB12 and CB2.3 The plant derived cannabinoids, Δ9-tetrahydrocannabinol (Δ9-THC), along with Δ8-THC and cannabidiol, were originally identified as having antineoplastic effects by Munson and colleagues in 1975.4 However, it is only relatively recently, since the receptors have been cloned and endogenous ligands been isolated, that there has been renewed interest in this field.
The first endocannabinoid to be isolated was N-arachidonoyl ethanolamine (anandamide, AEA).5 Anandamide and other endocannabinoids, including 2-arachidonoyl-glycerol, are present within the gastrointestinal tract6 and play a role in the control of many functions, including gastric motility.7–9 However, there is also accumulating evidence that endocannabinoids have the ability to modulate cell proliferation. Anandamide has been shown to have CB receptor mediated antitumour effects in a number of different tumour derived cell lines, including breast,10 mouse, and human lymphoblastic tumour cells,11 rat glioma,12 prostate,13 and cervical carcinoma cells.14 Data also suggest that endocannabinoids inhibit the proliferation of a colorectal cancer cell line (CaCo-2) via CB1 activation.15
Interestingly, as well as its cannabinoid receptor mediated actions, anandamide can be transported into the cell where it is either degraded into arachidonic acid and ethanolamine by fatty acid amide hydrolase (FAAH)16–18 or metabolised by cyclooxygenase 2 (COX-2) to prostaglandin-ethanolamides (PG-EAs).19,20 COX enzymes are also responsible for the conversion of arachidonic acid into prostaglandins (PGs) and thromboxane. COX-2 is overexpressed in the majority of colorectal cancers compared with normal epithelium,21,22 and evidence from clinical,23 animal,24,25 and in vitro studies26–28 have established that COX-2 is associated with the promotion of tumorigenesis. PGs, for example PGE2, are likely to mediate some of the tumour promoting effects of COX-2 as it has the ability to modulate the immune response to tumour cells,29 promote angiogenesis,30 and stimulate cell proliferation.31
As anandamide is growth inhibitory in some tumour types and can be metabolised by COX-2, our aim was to try to exploit the high levels of COX-2 found in colorectal tumour cells by targeting them for growth inhibition/cell death. Our hypothesis was that the growth inhibitory effects of anandamide, at least in part, could be through its metabolism by COX-2 and this would lead to specific growth inhibition/cell death in high COX-2 positive cells.
In the current study, we present data that show, for the first time, that anandamide induces cell death in COX-2 expressing colorectal carcinoma (CRC) cell lines. Cell death induced by anandamide in CRC cell lines was neither classical apoptosis nor necrosis, but was partially attenuated by the COX-2 selective inhibitors NS398 and rofecoxib. Furthermore, inhibition of FAAH activity (preventing degradation of anandamide into arachidonic acid and ethanolamine) potentiated anandamide induced cell death. The COX-2 dependent metabolites of anandamide, PGE2-EA and PGD2-EA, induced apoptosis in CRC cells, rather than non-apoptotic cell death. Taken together, the evidence presented here suggests that anandamide induces non-apoptotic cell death in COX-2 expressing CRC cell lines that is, at least in part, COX-2 dependent and could prove useful for specific targeting of COX-2 positive colorectal tumour cells.
METHODOLOGY
Materials
SW480 and HT29 were obtained from the American Type Culture Collection and HCA7/C29 were a kind gift from Susan Kirkland (Department of Histopathology, Imperial College London, UK). Anandamide (Sigma, St Louis, Missouri, USA) was prepared to a stock solution of 10 mM in absolute ethanol. MAFP was purchased from Tocris Cookson (Bristol, UK), hydrogen peroxide (H2O2) from BDH (Poole, Dorset, UK), and PGE2-ethanolamide and PGD2-ethanolamide were from Cayman Chemical (Tallinn, Estonia). TRAIL was a kind gift from Marion Macfarlane (MRC Toxicology Unit, University of Leicester, Leicester, UK).
Treatment of colorectal carcinoma cell lines
CRC cell lines were maintained in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum (FBS), glutamine (2 mM), penicillin (100 U/ml), and streptomycin (100 μg/ml) (standard growth medium) at 37°C in an atmosphere containing 5% CO2. For all treatment conditions, 2% FBS Dulbecco’s modified Eagle’s medium/F12 NUT mix medium containing glutamine (2 mM), penicillin (100 U/ml), and streptomycin (100 μg/ml) (2% NUT mix) was used in place of standard growth medium. All treatments were diluted in vehicle containing 2% NUT mix to give the indicated concentrations, such that each treatment condition contained the same amount of vehicle.
Cell lines were seeded in T25 flasks and after 72 hours of culture, cells were treated with appropriate concentrations of anandamide or vehicle control for 72 hours, at which point three day experiments were harvested. Adherent cells (those remaining attached to the flask) were removed by trypsinisation and shed cells (those having detached from the adherent monolayer) were counted separately. For six day experiments, the medium was removed from the flasks, after 72 hours, and replaced with fresh 2% NUT mix medium containing the appropriate concentration of anandamide or vehicle control. Shed cells were collected from the medium and counted at the 72 hour time point. After a total of six days of treatment, adherent and shed cells were counted separately and once again shed cells were represented as a percentage of the total cell yield, as previously described by Diaz and colleagues.32 For all experiments, each treatment condition and controls were conducted in triplicate and repeated at least three times.
Acridine orange and ethidium bromide staining
Fluorescence microscopy was used for the morphological detection of apoptotic cells, based on a method used by Gregory and colleagues33 and Hague and colleagues.34 Adherent and shed cells were stained with acridine orange (5 μg/ml) (Sigma) and ethidium bromide (5 μg/ml) (Sigma), which fluorescently label DNA. Acridine orange fluoresces yellow/green under light of excitation wavelength 450–490 nm and penetrates living cells whereas ethidium bromide can only enter permeable cells and stains red under light of the same excitation wavelength. Apoptotic cells were morphologically distinguished under a fluorescent microscope by their condensation and segregation of chromatin, cellular shrinkage, and formation of apoptotic bodies. Apoptotic/non-apoptotic cells were counted and calculated as a percentage of total adherent cells, as previously described.27
Western immunoblotting
Western blotting was carried out as described previously.35 COX-2 protein was detected using a mouse monoclonal COX-2 antibody (Cayman Chemical) at 1:500. COX-1 protein was detected using a goat polyclonal COX-1 antibody (Santa Cruz, California, USA) at 1:250. PARP was detected using a mouse monoclonal antibody (Alexis Corporation, Nottingham, UK) at 1:5000. FAAH protein was detected using a rabbit polyclonal FAAH antibody (Alexis Biochemicals) at 1:500. As a loading control, α-tubulin was detected using a mouse monoclonal (Sigma) at 1:10 000. The horseradish peroxidase (HRP) conjugated secondary antibody was goat antimouse (Sigma) at 1:1000; for the detection of FAAH, a HRP conjugated antirabbit antibody (Sigma) at 1:1000 and for the detection of COX-1 a HRP conjugated antigoat antibody (Sigma) was used at 1:1000.
Reverse transcriptase-polymerase chain reaction (RT-PCR)
Subconfluent cells were collected, and total RNA was isolated using an RNeasy mini kit (Qiagen, Crawley, West Sussex, UK). Following extraction, RNA was treated with DNase I (Ambion, Huntingdon, Cambridgeshire, UK). cDNA synthesis was carried out using 10 μg of total RNA in a 50 μl reaction containing 50 mM Tris HCl (pH 8.3), 75 mM KCl, 3 mM MgCl2, 10 mM DTT, 500 ng random hexamers, 1 μg oligo (dT)15 primer, 400 μM dNTPs, 40 units of RNase inhibitor, and 400 units of MMLV reverse transcriptase (Promega, Southampton, UK). FAAH transcripts were detected using the primers 5′-GAG GCT TCC GTG TCC TCT C-3′ (forward) and 5′-CCT ATG TCA TAC CCA TGG GC-3′ (reverse) to amplify a 138 bp product36; glyceraldehyde-3-phosphate dehydrogenase transcripts were detected using the primers 5′-CTT CAC CAC CAT GGA GAA GGC-3′ (forward) and 5′-GGC ATG GAC TGT GGT CAT GAG-3′ (reverse) to amplify a 238 bp product, as a control. PCR products were subjected to electrophoresis on a 2% agarose gel, and DNA was visualised with ethidium bromide staining.
Detection of nucleosomal fragmentation of genomic DNA
DNA extraction and electrophoresis on agarose gel were carried out as previously described.37 Both shed and adherent cells (1–2×106) were run on a 2% agarose gel (w/v) containing 0.1 μg/ml ethidium bromide. The gel was run at 40 V until the dye front had migrated 4–5 cm.
Labelling of externalised phosphatidylserine using annexin V-FITC
A modified technique for binding of annexin V-FITC and propidium iodide was used.38 Briefly, shed cells were harvested as previously described and 5×105 cell aliquots were resuspended in binding buffer (10 mM Hepes, 140 mM NaCl, 2.5 mM CaCl2, pH 7.4) and incubated at room temperature for 10 minutes with 10 μl annexin V-FITC (10 μg/ml) BD Bioscience Europe, Cowley, Oxford, UK. Following washing, cells were stained with 40 μl of propidium iodide 20 μg/ml before analysis by flow cytometry on a FACS Calibur flow cytometer using Cell Quest software (BD Bioscience Europe).
Adherent cells were washed with phosphate buffered saline and labelled with 25 μl annexinV-FITC (10 μg/ml) and 100 μl propidium iodide (20 μg/ml) in binding buffer and incubated for 10 minutes at room temperature. Plates were washed with binding buffer and adherent cells were harvested by scraping and resuspended at 5×105/ml for analysis.
PGE2 enzyme immunoassay
PGE2 and PGE2-EA were detected in culture medium using a PGE2 enzyme immunoassay (Cayman Chemical Co.). PGE2 enzyme immunoassay detects both PGE2 and PGE2-EA but cannot distinguish between them. Cells were seeded and treated with anandamide as described previously. After 72 hours of treatment, culture medium was collected, shed cells were removed, and aliquots of culture medium were assayed. The PGE2 assay was performed according to the manufacturer’s instructions. The immunoassay was sensitive to 15 pg/ml PGE2/PGE2-EA production. PGE2/PGE2-EA production was normalised for cell number and represented as pg/106 cells.
RESULTS
COX-2 expressing tumour cells are sensitive to anandamide induced cell death
The endogenous cannabinoid anandamide can be transported into the cell where it can be metabolised by cyclooxygenase enzymes into PG-EAs.19,20 As COX-2 is overexpressed in the majority of CRC, we sought to determine whether anandamide induced cell death in CRC cells and the possible COX-2 dependent effects of anandamide. To assess the response of CRC cells to anandamide, we used three human CRC cell lines with different basal COX-2 protein expression (fig 1A).

{kind=link}
{kind=link}
Figure 1
Cyclooxygenase (COX) expression and induction of cell death by anandamide in colorectal carcinoma (CRC) cell lines. (A) Sodium dodecyl sulphate-polyacrylamide gel western blot analysis of COX-1 and COX-2 protein levels in SW480, HT29, and HCA7/C29 cells. HCA7/C29 are known to express high levels of COX-2 protein39 and were used here as a positive control. SW480 express very low levels of COX-2 and in the exposure shown COX-2 protein is not visible in SW480 cells but could be detected in longer exposures. Equal loading was confirmed by reprobing for α-tubulin. (B) Three CRC cell lines were treated with vehicle control or anandamide 25 or 50 μM for a period of 72 hours. Adherent cell yields are represented as a percentage of vehicle control. Anandamide treatment resulted in decreased cell yield in HT29 and HCA7/C29 cells (moderate and high COX-2 expressors, respectively) with a corresponding increase in the proportion of cell shedding (C), representing cell death. The very low COX-2 expressor (SW480) was not affected by anandamide treatment. **p<0.01, ***p<0.001 versus control by Dunnett’s post hoc t test. All data are means (SEM) from three separate experiments, conducted in triplicate. Blank controls (2% fetal bovine serum (FBS) NUT mix medium) were also included but are not shown as there was no significant difference when compared with vehicle control (2% FBS NUT mix medium+ethanol) to show ethanol did not have any stimulatory or inhibitory effect on cell yield.
COX-2 expression was detected in all three cell lines (fig 1A), with HCA7/C29 cells expressing high levels of COX-239 compared with moderate levels expressed by HT29, in agreement with Crew and colleagues.40 SW480 expressed very low levels of COX-2 protein, as previously reported.41 Anandamide significantly reduced the adherent cell yield of COX-2 expressing HT29 and HCA7/C29 cells following a 72 hour treatment period (fig 1B). The decrease in cell yield was associated with induction of cell shedding (fig 1C), which is indicative of cell death.34 However, cell growth of the very low basal COX-2 expressors, SW480, was not significantly affected by anandamide treatment.
Selective inhibition of COX-2 enzyme activity attenuates anandamide induced cell death
As the response of CRC cells to anandamide correlated with basal COX-2 protein expression, we hypothesised that COX-2 enzyme activity may play an important role in the response of CRC cells to anandamide. NS398 is a COX-2 selective inhibitor and we have previously shown that 10 μM NS398 selectively inhibits COX-2 enzyme activity, as determined by a PGE2 enzyme immunoassay, without affecting cell survival.40 Therefore, CRC cells were pretreated with NS398 for 24 hours and then treated with anandamide, in the presence or absence of NS398 (fig 2A–F). For these experiments, we used 1, 10, and 25 μM anandamide over a longer time period (six days) to determine whether the differential sensitivity according to COX-2 expression was seen at lower doses.

{kind=link}
{kind=link}
Figure 2
Anandamide (AEA) induced cell death was partially attenuated by the cyclooxygenase 2 (COX-2) selective inhibitor NS398. Three colorectal carcinoma cell lines, SW480 (A, B), HT29 (C, D) and HCA7/C29 (E, F) were pretreated with 10 μM NS398 for 24 hours followed by treatment with AEA or with AEA alone. Cells were retreated after 72 hours when cells shed into the medium were counted. After a total of six days of treatment with AEA, adherent and shed cells were harvested, based on a method by Diaz and colleagues.32 Adherent cell yield was represented as a percentage of vehicle control (A, C, E). Shed cell numbers were pooled at the 72 hour and six day time points and represented as a proportion of total cell yield (B, D, F). One way ANOVA indicated that NS398 significantly protected against AEA induced cell death in HT29 cells at all three concentrations of AEA, for both adherent (p<0.05) and shed cell data (p<0.001), and HCA7/C29 cells for both adherent and shed cell data (p<0.05) at 10 μM AEA and above. NS398 did not affect the response of SW480 cells to AEA. Data are means (SEM) from three separate experiments conducted in triplicate. Controls included vehicle control, and NS398; for all cell lines there was no significant difference between vehicle and NS398 treated cells.
Interestingly, even after six days of treatment, anandamide did not induce significant cell death in the low COX-2 expressing cell line, SW480, either alone or in combination with NS398 (fig 2A, B). However, anandamide reduced adherent cell yield in COX-2 expressing cell lines HT29 and HCA7/C29 (fig 2C, E, respectively) with a corresponding increase in shed cell yield (fig 2D, F). Selective inhibition of COX-2 significantly (p<0.05) protected against cell death induced by anandamide in HT29 at all concentrations of anandamide, and HCA7/C29 at 10 μM and above (p<0.05). Similar results were obtained with another COX-2 selective inhibitor, rofecoxib, where cell death induced by anandamide in HT29 was significantly reduced by rofecoxib (data not shown).
Cell death induced by anandamide
The antiproliferative effects of anandamide have largely been attributed to cell cycle arrest10 or induction of apoptosis,11,12,14,42,43 but induction of cell death in CRC cells has yet to be investigated. Therefore, due to the increase in cell shedding, anandamide treated cell samples were analysed for apoptotic and necrotic morphology following acridine orange and ethidium bromide staining. Spontaneous apoptosis occurs in CRC cells at a rate of 5–15% depending on the cell line,44 and therefore vehicle control shed cell samples are used as a positive control for apoptosis. Apoptotic morphology includes membrane blebbing, condensation and segregation of nuclear chromatin, and cellular shrinkage, demonstrated in vehicle control treated samples (fig 3A). Unlike apoptosis, necrosis involves cellular swelling, disruption of organelles, and loss of plasma membrane integrity (fig 3C). Interestingly, anandamide treated cells were predominantly stained with ethidium bromide, displayed cellular shrinkage, membrane blebbing, and chromatin condensation (fig 3B), but chromatin did not become segregated even after six days of treatment (data not shown).

{kind=link}
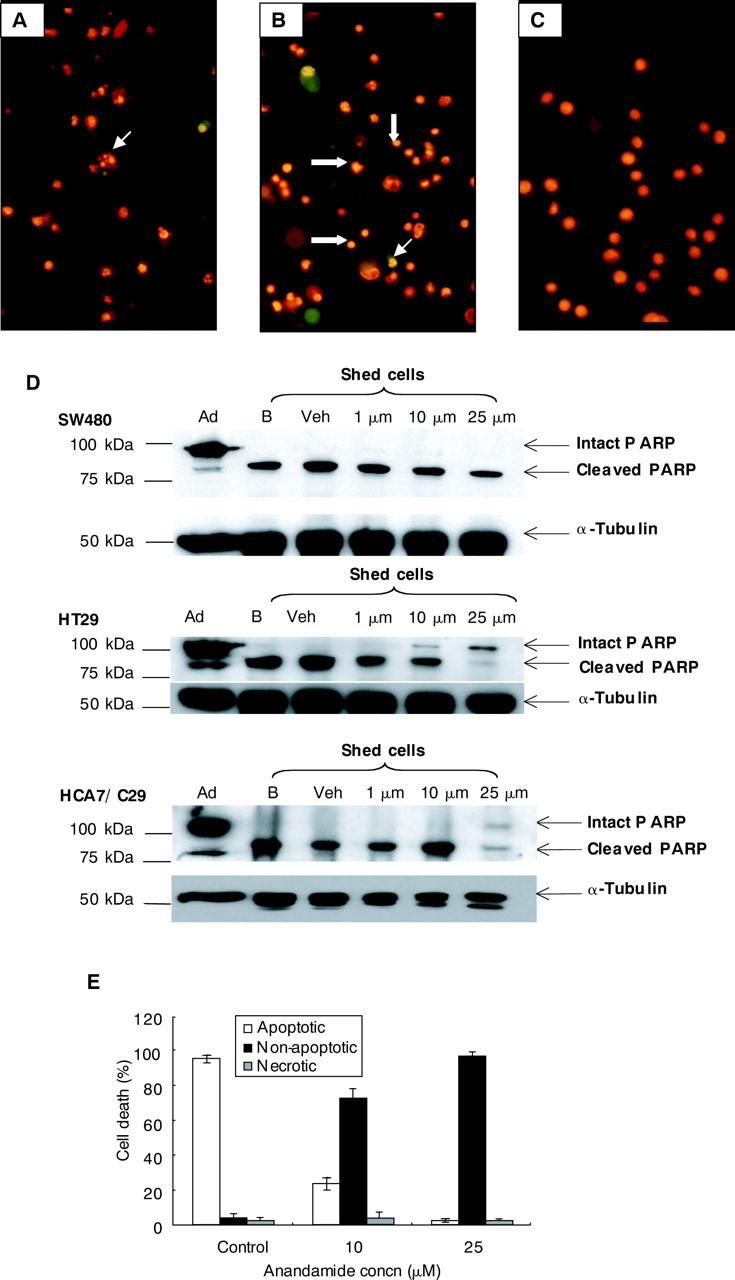{kind=link}
Figure 3
Determination of anandamide induced cell death. Shed cells were collected from control and anandamide treated cells and were dual stained with acridine orange (AO) (5 μg/ml) and ethidium bromide (EB) (5 μg/ml). Spontaneous apoptosis was evident in vehicle control cells (A); arrow indicates apoptotic cells with condensation and segregation of chromatin, membrane blebbing, and cellular shrinkage. Early apoptotic cells stain green and late apoptosis/secondary necrosis fluoresce red. (B) HT29 cells treated with 25 μM anandamide (six days) and stained with AO/EB. Anandamide treated cells were predominantly stained with EB, were small in size, and the chromatin had condensed but did not segregate. Examples are indicated with block arrows; apoptosis was also detected in anandamide treated samples (fine arrow) at a rate of ∼1%. (C) H2O2 (72 hours) induced necrosis in HT29 cells, represented by the uptake of EB, with swollen nuclei present in all cells within the field. (D) SW480, HT29, and HCA7/C29 cells were treated for six days with anandamide, and adherent and shed cell lysates were collected from experiments shown in fig 2. Intact (116 kDa) and cleaved PARP (89 kDa) were detected in adherent cell samples (Ad) and cleaved PARP was detected in blank (B; 2% fetal bovine serum (FBS) NUT mix medium) and vehicle control shed cell samples (Veh; 2% FBS NUT mix medium+0.25% ethanol) due to spontaneous apoptosis in colorectal carcinoma cell lines. Anandamide induced significant cell death in both HT29 and HCA7/C29 at 10 μM anandamide and above, which corresponds to a decrease in the amount of cleaved PARP and the appearance of intact PARP (116 kDa). Repeat probing for α-tubulin controls for equal loading and transfer. (E) HT29 cells were treated with vehicle control (Control), or 10 μM or 25 μM anandamide for a total of six days. Shed cells were collected and stained with AO and EB, and 300 cells from randomly selected fields were counted and the proportion of cells with apoptotic, non-apoptotic, and necrotic morphology were calculated.
We then investigated the presence/absence of biochemical markers of apoptosis. In all adherent cell samples the predominant form of PARP was intact (116 kDa) but small amounts of cleaved (89 kDa) PARP were also detected due to spontaneous apoptosis in the adherent monolayer (fig 3D). In SW480, HT29, and HCA7/C29 blank (B) and vehicle (Veh) control shed cell samples, only cleaved PARP was detected, suggesting that all of these cells had undergone apoptosis (fig 3D). Following six days of treatment with anandamide, SW480 shed cells also expressed cleaved PARP, which could be attributed to spontaneous apoptosis (fig 3D) as there was no significant induction of shed cells in this cell line (see fig 2B). Interestingly, concentrations of anandamide that induced significant cell shedding in HT29 and HCA7/C29 cells (fig 2D, 2F), corresponded with a reduction in cleaved PARP at 10 μM, and this effect was more pronounced with 25 μM anandamide with the appearance of intact PARP in the shed cells (fig 3D). The reduction of cleaved and increase in intact PARP corresponded to a decrease in cells with an apoptotic morphology and an increase in a proportion of cells with non-apoptotic morphology (fig 3D, E), while the proportion of morphologically necrotic cells did not change between control and anandamide treated samples (fig 3E). Therefore, when comparing expression of cleaved and intact PARP in similar numbers of treated and untreated shed cells with shed cell morphology, 10 and 25 μM anandamide shifted the balance from apoptotic to non-apoptotic cell death in COX-2 expressing cell lines. Furthermore, death induced by anandamide was caspase independent as the caspase inhibitor z-VAD-fmk did not protect against this form of non-apoptotic cell death (data not shown).
Internucleosomal DNA fragmentation is one of the final events during the execution phase of apoptosis. Spontaneous cell shedding and TRAIL treatment represent positive controls for apoptosis, producing a DNA ladder, whereas H2O2 treatment was used as a positive control for necrosis45 resulting in non-specific DNA fragmentation shown as a high molecular weight band and a smear (fig 4A). We used a concentration of anandamide (25 μM) that we have previously shown to induce significant cell death that does not appear to be apoptosis. Interestingly, anandamide treated cells did not produce internucleosomal DNA ladder, and instead non-specific DNA fragmentation was detected that was similar to necrosis (fig 4A).

{kind=link}
{kind=link}
Figure 4
Anandamide induced cell death was neither apoptosis nor necrosis. (A) HT29 cells were treated for six days with either vehicle control (control) or 25 μM anandamide (anandamide) and adherent (Ad) and shed (F) cell samples were subjected to DNA extraction and run on an agarose gel. HT29 cells were also treated with 0.5 μg/ml TRAIL for 16 hours as a positive control for apoptosis or 32 mM H2O2 as a positive control for necrosis. The pattern of DNA fragmentation for anandamide treated cells appeared to be different from apoptosis but produced a similar pattern to necrosis. (B) Dual parameter blots of propidium iodide (PI) versus annexin V-FITC obtained in (i) vehicle control HT29 shed cells (Spontaneous), (ii) 25 μM anandamide shed cells (Anandamide), or (iii) 32 mM H2O2 shed cells (H2O2) treated for 72 hours. Percentages shown are means (SD) of proportions of cells distributed within quadrants from at least three separate experiments. Lower left, negative; lower right, annexin V; upper right, annexin V/PI; upper left, PI.
As a further test for characterisation of anandamide induced cell death, we investigated annexin V binding. Annexin V staining is used as a marker for apoptosis38 and in combination with propidium iodide can be used for distinguishing between apoptotic and necrotic cells. As expected in spontaneously shed cell samples, it was possible to distinguish early apoptosis (annexin V positive), late apoptosis/secondary necrosis (annexin V and propidium iodide (PI positive)), necrosis (PI positive), and those cells that are negative for both annexin V and PI (fig 4B). Annexin V positive cells were also detected in anandamide treated shed cell samples but the majority of these also stained positive for both annexin V and PI (mean 51.2 (SD 8.5)%). However, there were significantly less cells positive for PI only (p<0.05) in anandamide treated cells (mean 13 (SD 3.1)%) compared with necrotic shed cells (H2O2) (39.3 (8.0)%) (fig 4B). Even after six days of treatment with anandamide, these cells remained positive for both annexin V and PI, with no increase in the proportion of PI only positive cells (data not shown). Taken together these results suggest that early stages of anandamide induced cell death appear to be similar to apoptosis in terms of phosphatidylserine exposure but the terminal stages are quite different, as indicated by cell morphology, PARP cleavage, and DNA ladder. Furthermore, analysis of anandamide treated cells by transmission electron microscopy confirmed that anandamide treated cells were not dying by classical apoptosis (data not shown). Even though anandamide induced cell death produced similar DNA fragmentation profiles to necrosis, the cell morphology and phosphatidylserine exposure were distinctly different to necrosis. Therefore, anandamide induced cell death did not appear to be either classical apoptosis or necrosis, and was classified as non-apoptotic cell death.
Anandamide increases PGE2-EA and/or PGE2 production
To determine whether anandamide treatment increases PG-EA production, we used a PGE2 enzyme immunoassay which detects both PGE2 and PGE2-EA but cannot distinguish between them. Total PGE2/PGE2-EA production was significantly (p<0.001) increased in HT29 cells following 72 hours of treatment with 25 μM anandamide (fig 5A). The increase in PGE2/PGE2-EA production could either be due to degradation of anandamide into arachidonic acid consequently providing greater substrate for COX-2 in the production of PGE2, or to direct metabolism of anandamide by COX-2 into PGE2-EA. Previous reports have suggested that treatment of cells with a stable analogue of anandamide, methanandamide, increased COX-2 mRNA and protein expression.46–48 Notably, anandamide did not increase COX-2 levels (fig 5B) and therefore the increase in PGE2/PGE2-EA production seen here was not due to upregulation of COX-2 protein expression but instead was likely due to the availability of the substrate.

{kind=link}
{kind=link}
Figure 5
Anandamide induced cell death corresponded to an increase in prostaglandin E2/ prostaglandin E2-ethanolamide (PGE2/PGE2-EA) secretion but did not affect cyclooxygenase 2 (COX-2) protein expression. (A) HT29 cells were treated with vehicle control (control) or the concentration of anandamide indicated, for 72 hours. PGE2/PGE2-EA was secreted into the medium and assessed using a PGE2 enzyme immunoassay (Cayman Chemical) which also detects PGE2-EA, but cannot distinguish between them. Anandamide 25 μM significantly increased PGE2/PGE2-EA secretion (***p<0.001). The lower limit of accurate detection of the assay was 15 pg/ml; †concentrations below this limit. The results shown are means of duplicate measurements. Similar results were obtained in repeat experiments. (B) Adherent cells from the above experiments were collected as cell lysates and subjected to sodium dodecyl sulphate-polyacrylamide gel immunoblotting for COX-2 protein expression. HCA7/C29 cells were used as a positive control for COX-2 protein expression (+ve), vehicle control (control), 1 μM anandamide (1 μM), 10 μM anandamide (10 μM), and 25 μM anandamide (25 μM). COX-2 protein expression was not affected by anandamide treatment over 72 hours (or six days, data not shown). Repeat probing for α-tubulin controls for equal loading and transfer.
Anandamide induced cell death is potentiated by inhibition of FAAH
Once inside the cell, anandamide can either be metabolised by COX-219,20 or degraded by the endogenous enzyme FAAH into arachidonic acid and ethanolamine. The increase in PGE2/PGE2-EA levels (fig 5A) seen during anandamide induced non-apoptotic cell death could therefore be mediated via FAAH degradation of anandamide into arachidonic acid and ethanolamine (and subsequent conversion of arachidonic acid into PGE2), or alternatively, through COX-2 metabolism of anandamide into PGE2-EA. We hypothesised that the increase in total PGE2/PGE2-EA production was likely due to an increase in PG-EA production as increasing arachidonic acid levels could potentially antagonise anandamide induced cell death as there would be a greater substrate for COX-2 dependent production of the growth promoting PGs.
Following confirmation of FAAH expression in our cell lines by RT-PCR and western blotting (fig 6A, B), we investigated whether inhibition of FAAH activity could potentiate the response to anandamide using the selective inhibitor MAFP.49 Treatment with 0.1 μM MAFP resulted in significant potentiation (p<0.001) of anandamide induced cell death in HT29 cells (fig 6C, D). Cell death induced by the combination of anandamide and MAFP did not appear to be apoptosis. Again there was a shift from cleaved PARP to intact PARP in shed cells (indicative of non-apoptotic cell death), with a greater decrease in the ratio of cleaved to intact PARP when anandamide was used in combination with MAFP (fig 6E). Similarly, shed cell morphology was identical to that of anandamide alone (data not shown). These data suggest that inhibition of FAAH activity potentiated non-apoptotic cell death induced by anandamide. Similar significant potentiation (p<0.001) of anandamide induced cell death by MAFP was also seen in HCA7/C29 cells (data not shown). Furthermore, treatment of CRC cells with either arachidonic acid or ethanolamine did not result in non-apoptotic cell death (data not shown). Therefore, as inhibition of COX-2 attenuates the response to anandamide induced non-apoptotic cell death (fig 2) and inhibition of FAAH activity by MAFP potentiates non-apoptotic cell death induced by anandamide (fig 6C–E), this suggests that anandamide induced cell death is likely due to metabolism of anandamide by COX-2, rather than degradation into arachidonic acid and ethanolamine.

{kind=link}
{kind=link}
Figure 6
Inhibition of anandamide hydrolysis to arachidonic acid via inhibition of fatty acid amide hydrolase (FAAH) potentiated anandamide induced cell death. (A) FAAH mRNA expression was detected in colorectal carcinoma (CRC) cell lines by reverse transcriptase-polymerase chain reaction. Lane 1, CaCo2 cells are known to express FAAH mRNA15; lane 2, SW480; lane 3, HT29; lane 4, HCA7/C29. The housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is shown in the lower panel. FAAH amplification was confirmed by sequencing. FAAH protein expression was confirmed by western blotting (B). The selective FAAH inhibitor MAFP significantly potentiated both growth inhibition induced by anandamide (AEA 25 μM) (C) and cell death (D) in HT29 cells after 72 hours of treatment. ***p<0.001 versus control by Dunnett’s post hoc t test. Data points shown are means (SEM) from three separate experiments conducted in triplicate. Shed cells were collected from these experiments and subjected to sodium dodecyl sulphate-polyacrylamide gel electrophoresis for the presence of cleaved PARP (E). Adherent cells samples (Ad) were used as a positive control for intact (116 kDa) and cleaved PARP (89 kDa). PARP cleavage was investigated in shed cell samples, blank control (blank) (2% FBS NUT mix medium), vehicle control (control) (2% FBS NUT mix medium+ethanol), 0.1 μM MAFP (MAFP), 25 μM anandamide (AEA), and 0.1 μM MAFP in combination with 25 μM anandamide (MAFP+AEA). Intact PARP was detected in both AEA alone and in the combination of AEA and MAFP but cell death induced by the combination of AEA and MAFP potentiated the reduction in PARP cleavage resulting in less cleaved PARP compared with anandamide only treated samples. Repeat probing for α-tubulin controls for equal loading and transfer.
PG-EA metabolites of anandamide induce apoptotic cell death in CRC cell lines
Anandamide induces non-apoptotic cell death which is, in part, COX-2 dependent. To determine whether the non-apoptotic cell death induced by anandamide was due to COX-2 dependent metabolites of anandamide, we investigated the response of CRC cell lines to PG-EAs. Although there is much literature on PGs having growth promoting properties, very little is known about the role of PG-EAs in growth regulation. Initial reports suggest PGE2-EA can interact with PGE2 receptors, EP1–4.50 However, to the best of our knowledge, the effect of PG-EAs on cancer cell growth has yet to be investigated and since data presented suggests PG-EAs may mediate the growth inhibitory affects of anandamide, it was important to study these molecules.
PGE2-EA reduced cell growth of CRC cell lines (fig 7A) by causing an increase in cell death (fig 7B). This is an interesting observation as it has previously been shown that similar concentrations of PGE2 cause a stimulatory effect on cell proliferation of CRC cells via activation of EP4.51,52 (and own unpublished data). Furthermore, PGD2-EA also reduced cell growth (fig 7C) and induced cell death in CRC cell lines (fig 7D). Analysis of shed cell morphology (data not shown), annexin V binding (data not shown), and PARP cleavage (fig 7E) revealed that unlike anandamide induced cell death, apoptosis was the major form of cell death produced by both PGE2-EA and PGD2-EA. In contrast PGF2α-EA treatment did not affect CRC cell growth (data not shown). This suggests that anandamide induced non-apoptotic cell death is not solely due to the production of a single PG-EA (either PGE2-EA, PGD2-EA, or PGF2α-EA) but most likely due to a combination of COX-2 dependent metabolites.

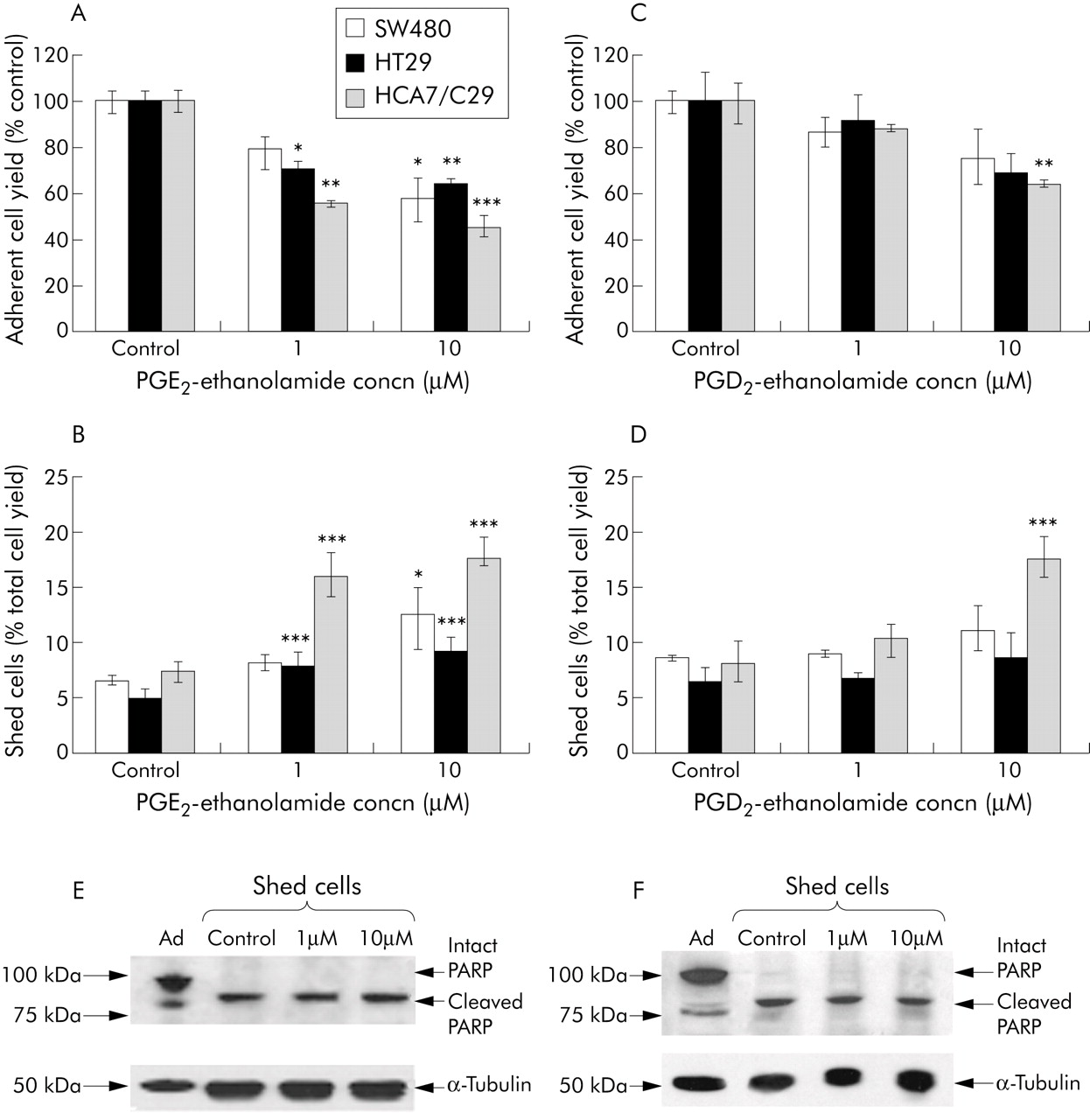{kind=link}
{kind=link}
Figure 7
Cyclooxygenase 2 (COX-2) metabolites of anandamide induced apoptosis in colorectal carcinoma cell lines. SW480, HT29, and HCA7/C29 cells were treated with the indicated concentrations of prostaglandin E2 (PGE2)-ethanolamide (A, B) and PGD2-ethanolamide (C, D) for 72 hours. Adherent cell yields are presented as a percentage of vehicle control (A, C). Shed cells are presented as a proportion of total cell yield (B, D). PGE2-ethanolamide and PGD2-ethanolamide inhibited cell growth and induced cell death in colorectal carcinoma cells. *p<0.05, **p<0.01, ***p<0.001 versus control, by Dunnett’s post hoc t test. All data are means (SEM) from three separate experiments, conducted in triplicate. Shed cells from these experiments were collected and subjected to sodium dodecyl sulphate-polyacrylamide gel electrophoresis for the presence of cleaved PARP. The example shown is from HT29 cells treated with PGE2-ethanolamide (E) and PGD2-ethanolamide (F). As a positive control for intact and cleaved PARP adherent cells treated with vehicle control were included (Ad), and vehicle control shed cell samples (Control) were included as a positive control for cleaved PARP.
DISCUSSION
Interest in the antiproliferative effects of endocannabinoids, particularly anandamide, stemmed from their ability to inhibit the growth of tumour cells, which can be dependent11,12,15 and independent42,50,53 of cannabinoid receptor activation. We were particularly interested in the CB receptor independent effects of anandamide as COX-2 is overexpressed in a high proportion of colorectal carcinomas,22 and anandamide can be metabolised by COX-2 in CRC cells.19,20 In this study, we investigated, for the first time, whether the endogenous cannabinoid anandamide and PG-EAs could induce cell death in CRC cell lines, whether the response to anandamide was dependent on COX-2, and if so whether we could exploit the high levels of COX-2 found in tumour cells to target them for cell death.
Anandamide significantly inhibited tumour cell growth and induced cell death in COX-2 expressing cell lines (HT29 and HCA7/29) while having no significant effect on the very low COX-2 expressing cell line (SW480). Having shown that anandamide induced cell death was greatest in high COX-2 expressing cells, we used the selective COX-2 inhibitor, NS398, in combination with anandamide to show that cell death was in part mediated by COX-2. Selective inhibition of COX-2 enzyme activity by NS398 significantly protected against anandamide induced growth inhibition and cell death in HT29 and HCA7/C29 cell lines. Therefore, unlike signalling in neurones, where inhibition of COX-2 metabolism of anandamide prolongs the response to anandamide,54 the antiproliferative effect of anandamide in CRC cell lines could be mediated via metabolism by COX-2. The fact that anandamide induces cell death in CRC cell lines in a manner that can be partially rescued by COX-2 inhibitors suggests that COX-2 metabolism of anandamide is, at least in part, responsible for the induction of cell death.
We presented evidence that anandamide treatment resulted in an increase in PGE2/PGE2-EA production. The increase in total PGE2/PGE2-EA production was unlikely to be attributable to an increase in PGE2 because rather than growth inhibitory effects, PGE2 is growth stimulatory in CRC cells31 (and own unpublished observations). Therefore, as COX-2 dependent metabolism of anandamide is growth inhibitory, which correlates with an increase in PGE2/PGE2-EA, this suggests that COX-2 metabolites of anandamide could be responsible for anandamide induced cell death. Furthermore, blocking FAAH activity in combination with anandamide treatment potentiated the non-apoptotic cell death seen when treating with anandamide alone. These results are in agreement with Maccarrone and colleagues42 and Fowler and colleagues,55 who also reported that the antiproliferative effects of anandamide were not mediated by arachidonic acid. Interestingly, a recent report also suggested that inhibition of endocannabinoid hydrolysis (via inhibition of FAAH) resulted in reduced growth of thyroid tumour cells subcutaneously transplanted into athymic mice.56
We found that anandamide induced non-apoptotic cell death. Interestingly, an unusual form of cell death has also been reported by Mimeault and colleagues13 in response to similar concentrations of anandamide in prostate carcinoma cells but was simply described as apoptotic/necrotic cell death. In a more recent study, using similar concentrations of anandamide, caspase activation was found to be unnecessary and was secondary to calpain activation in mediating apoptosis via CB2 activation.53 Similar cell morphology and lack of PARP cleavage has also been reported in response to activation of PPARγ,56 and absence of DNA laddering was detected following irradiation of breast carcinoma cells.45 Induction of cell death other than apoptosis could be particularly beneficial for those tumour cells that have become resistant to induction of apoptosis, and currently there is growing interest in the field of non-apoptotic forms of cell death.57
As the data suggest that COX-2 mediates, at least in part, the cytotoxic effects of anandamide, we investigated whether any of the known COX-2 metabolites of anandamide were growth inhibitory. Both PGE2-EA and PGD2-EA were growth inhibitory. This is in direct contrast with PGE2, which is growth stimulatory51,52 (and own unpublished data). Interestingly, although cell death induced by anandamide is neither apoptosis nor necrosis, both PGE2-EA and PGD2-EA induced classical apoptosis, suggesting that induction of cell death by anandamide may involve other (COX-2) metabolites, for example 11-hydroxy-eicosatetraenoic acid and 15-hydroxy-eicosatetraenoic acid,58 and/or a combination of factors, including PG-EAs.
In summary, this is the first report demonstrating that anandamide induces cell death in COX-2 expressing colorectal tumour cells and interestingly, that COX-2 dependent metabolites of anandamide, PG-EAs, induce apoptosis in CRC cells. Non-apoptotic cell death induced by anandamide in CRC cells is, at least in part, COX-2 dependent. This raises the exciting possibility that high COX-2 expressing colorectal tumour cells can be targeted for cell death by anandamide while sparing normal cells which do not express COX-2. As COX-2 is overexpressed in a number of tumour types, as well as in the majority of colorectal cancers, it would be interesting to examine whether high COX-2 expressing tumour cells from other cancer types would respond to endogenous cannabinoid treatment. Also, the use of agents such as anandamide may prove to be of particular benefit in treating tumours which have become resistant to apoptosis.
Acknowledgments
This work was supported by a programme grant from Cancer Research UK and the Citrina Foundation.
REFERENCES
- ↵ Guzman M. Cannabinoids: potential anticancer agents. Nat Rev Cancer2003;3:745–55.CrossRefPubMedWeb of ScienceGoogle Scholar
- ↵ Matsuda LA, Lolait SJ, Brownstein MJ, et al. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature1990;346:561–5.CrossRefPubMedWeb of ScienceGoogle Scholar
- ↵ Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature1993;365:61–5.CrossRefPubMedWeb of ScienceGoogle Scholar
- ↵ Munson AE, Harris LS, Friedman MA, et al. Antineoplastic activity of cannabinoids. J Natl Cancer Inst1975;55:597–602.Google Scholar
- ↵ Devane WA, Hanus L, Breuer A, et al. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science1992;258:1946–9.Abstract/FREE Full TextGoogle Scholar
- ↵ Di Carlo G, Izzo AA. Cannabinoids for gastrointestinal disease: potential therapeutic applications. Expert Opin Investig Drugs2003;12:39–49.PubMedWeb of ScienceGoogle Scholar
- ↵ Calignano A, La Rana G, Makriyannis A, et al. Inhibition of intestinal motility by anandamide, an endogenous cannabinoid. Eur J Pharmacol1997;340:R7–8.CrossRefPubMedWeb of ScienceGoogle Scholar
- Izzo AA, Mascolo N, Capasso F. The gastrointestinal pharmacology of cannabinoids. Curr Opin Pharmacol2001;1:597–603.CrossRefPubMedGoogle Scholar
- ↵ Pinto L, Izzo AO, Cascio MG, et al. Endocannabinoids as physiological regulators of colonic propulsion in mice. Gastroenterology2002;123:227–34.CrossRefPubMedWeb of ScienceGoogle Scholar
- ↵ De Petrocellis L, Melck D, Palmisano A, et al. The endogenous cannabinoid anandamide inhibits human breast cancer cell proliferation. Proc Natl Acad Sci USA1998;95:8375–80.Abstract/FREE Full TextGoogle Scholar
- ↵ McKallip RJ, Lombard C, Fisher M, et al. Targeting CB2 cannabinoid receptors as a novel therapy to treat malignant lymphoblastic disease. Blood2002;100:627–34.Abstract/FREE Full TextGoogle Scholar
- ↵ Jacobsson SOP, Wallin T, Fowler CJ. Inhibition of rat C6 glioma cell proliferation by endogenous and synthetic cannabinoids: relative involvement of cannabinoid and vanilloid receptors. J Pharmacol Exp Ther2001;299:951–9.Abstract/FREE Full TextGoogle Scholar
- ↵ Mimeault M, Pommery N, Wattez N, et al. Anti-proliferative and apoptotic effects of anandamide in human prostatic cancer cell lines: Implication of epidermal growth factor receptor down-regulation and ceramide production. Prostate2003;56:1–12.CrossRefPubMedWeb of ScienceGoogle Scholar
- ↵ Contassot E, Tenan M, Schnuriger V, et al. Arachidonyl ethanolamide induces apoptosis of uterine cervix carcinoma cells via aberrantly expressed vanilloid receptors-1. Gynecol Oncol2004;93:182–8.CrossRefPubMedGoogle Scholar
- ↵ Ligresti A, Bisogno T, Matias I, et al. Possible endocannabinoid control of colorectal cancer cell growth. Gastroenterology2003;125:677–87.CrossRefPubMedWeb of ScienceGoogle Scholar
- ↵ Deutsch DG, Chin SA. Enzymatic synthesis and degradation of anandamide, a cannabinoid receptor agonist. Biochem Pharmacol1993;46:791–6.CrossRefPubMedWeb of ScienceGoogle Scholar
- Cravatt BF, Giang DK, Mayfield SP, et al. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature1996;384:83–7.CrossRefPubMedWeb of ScienceGoogle Scholar
- ↵ Maccarrone M, van der Stelt M, Rossi A, et al. Anandamide hydrolysis by human cells in culture and brain. J Biol Chem1998;273:32332–9.Abstract/FREE Full TextGoogle Scholar
- ↵ Yu M, Ives D, Ramesha CS. Synthesis of prostaglandin E2 ethanolamide from anandamide by cyclooxygenase-2. J Biol Chem1997;272:21181–6.Abstract/FREE Full TextGoogle Scholar
- ↵ Kozak KR, Crews BC, Morrows JD, et al. Metabolism of the endocannabinoids, 2-arachidonylglycerol and anandamide, into prostaglandin, thromboxane, and prostacyclin glycerol esters and ethanolamides. J Biol Chem2002;277:44877–85.Abstract/FREE Full TextGoogle Scholar
- ↵ Eberhart CE, Coffey RJ, Radhika A, et al. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology1994;107:1183–8.PubMedWeb of ScienceGoogle Scholar
- ↵ Elder DJ, Baker JA, Banu NA, et al. Human colorectal adenomas demonstrate a size-dependent increase in epithelial cyclooxygenase-2 expression. J Pathol2002;198:428–34.CrossRefPubMedWeb of ScienceGoogle Scholar
- ↵ Kune GA, Kune S, Watson LF. Colorectal cancer risk, chronic illnesses, operations and medications: a case control results from the Melbourne colorectal cancer study. Cancer Res1988;48:4399–404.Abstract/FREE Full TextGoogle Scholar
- ↵ Oshima M, Dinchuk JE, Kargman SL, et al. Suppression of intestinal polyposis in ApcΔ716 knockout mice by inhibition of cyclo-oxygenase-2 (COX-2). Cell1996;87:803–9.CrossRefPubMedWeb of ScienceGoogle Scholar
- ��� Jacoby RF, Seibert K, Cole CE, et al. The cyclooxygenase-2 inhibitor celecoxib is a potent preventive and therapeutic agent in the min mouse model of adenomatous polyposis. Cancer Res2000;60:5040–4.Abstract/FREE Full TextGoogle Scholar
- ↵ Tsujii M, Dubois RN. Alterations in cellular adhesion and apoptosis in epithelial cell overexpressing prostaglandin endoperoxide synthase 2. Cell1995;83:493–501.CrossRefPubMedWeb of ScienceGoogle Scholar
- ↵ Elder DJE, Hague A, Hicks DJ, et al. Differential growth inhibition by the aspirin metabolite salicylate in human colorectal tumour cell lines: enhanced apoptosis in carcinoma and in vitro-transformed adenoma relative to adenoma cell lines. Cancer Res1996;56:2273–6.Abstract/FREE Full TextGoogle Scholar
- ↵ Tsujii M, Kawano S, Dubois RN. Cyclooxygenase-2 expression in human colon cancer cells increases metastatic potential. Proc Natl Acad Sci USA1997;94:3336–40.Abstract/FREE Full TextGoogle Scholar
- ↵ Harris SG, Padilla J, Koumas L, et al. Prostaglandins as modulators of immunity. Trends Immunol2002;23:144–50.CrossRefPubMedWeb of ScienceGoogle Scholar
- ↵ Tsujii M, Kawano S, Tsujii S, et al. Cyclooxygenase regulated angiogenesis induced by colon cancer cells. Cell1998;93:705–16.CrossRefPubMedWeb of ScienceGoogle Scholar
- ↵ Qiao L, Kozoni V, Tsiolias GJ, et al. Selected eicosanoids increase the proliferation rate of human colon carcinoma cell lines and mouse colonocytes in vivo. Biochem Biophys Acta1995;1258:215–23.PubMedWeb of ScienceGoogle Scholar
- ↵ Diaz GD, Paraskeva C, Thomas MG, et al. Apoptosis is induced by the active metabolite of vitamin D3 and analogue EB1089 in colorectal adenoma and carcinoma cells: possible implications for prevention and therapy. Cancer Res2000;60:2304–12.Abstract/FREE Full TextGoogle Scholar
- ↵ Gregory CD, Dive C, Henderson S, et al. Activation of Epstein-Barr virus latent genes protects human B cells from death by apoptosis. Nature1991;349:612–14.CrossRefPubMedWeb of ScienceGoogle Scholar
- ↵ Hague A, Manning AM, Hanlon KA, et al. Sodium butyrate induces apoptosis in human colonic tumour cell lines in a p53-independent pathway: implications for the possible role of dietary fiber in the prevention of large-bowel cancer. Int J Cancer1993;55:498–505.CrossRefPubMedWeb of ScienceGoogle Scholar
- ↵ Elder DJE, Halton DE, Crew TE. et al Apoptosis induction and cyclooxygenase-2 regulation in human colorectal adenoma and carcinoma cell lines by the cyclooxygenase-2-selective non-steroidal anti-inflammatory drug NS-398. Int J Cancer2000;86:553–60.CrossRefPubMedWeb of ScienceGoogle Scholar
- ↵ Wan M, Cravatt BF, Ring HZ, et al. Conserved chromosomal location and genomic structure of human and mouse fatty-acid amide hydrolase genes and evaluation of clasper as a candidate neurological mutation. Genomics1998;54:408–14.CrossRefPubMedWeb of ScienceGoogle Scholar
- ↵ Smith CA, Williams GT, Kingston R, et al. Antibodies to CD3/T-cell receptor complex induce death by apoptosis in immature T cells in thymic cultures. Nature1989;337:181–4.CrossRefPubMedGoogle Scholar
- ↵ Clarke RG, Lund EK, Johnson IT, et al. Apoptosis can be detected in attached colonic adenocarcinoma HT29 cells using annexin V binding, but not by TUNEL assay of Sub-G0 DNA content. Cytometry2000;39:141–50.CrossRefPubMedWeb of ScienceGoogle Scholar
- ↵ Sheng H, Shao J, Kirkland SC, et al. Inhibition of human colon cancer cell growth by selective inhibition of cyclooxygenase-2. J Clin Invest1997;99:2254–9.CrossRefPubMedWeb of ScienceGoogle Scholar
- ↵ Crew TE, Elder DJE, Paraskeva C. A cyclooxygenase-2 (COX-2) selective non-steroidal anti-inflammatory drug enhances the growth inhibitory effect of butyrate in colorectal carcinoma cells expressing COX-2 protein: regulation of COX-2 by butyrate. Carcinogenesis2000;21:69–77.Abstract/FREE Full TextGoogle Scholar
- ↵ Richter M, Weiss M, Weinberger I, et al. Growth inhibition and induction of apoptosis in colorectal tumour cells by cyclooxygenase inhibitors. Carcinogenesis2001;22:17–25.Abstract/FREE Full TextGoogle Scholar
- ↵ Maccarrone M, Lorenzon T, Bari M, et al. Anandamide induces apoptosis in human cells via vanilloid receptors. Evidence for a protective role of cannabinoid receptors. J Biol Chem2000;275:31938–45.Abstract/FREE Full TextGoogle Scholar
- ↵ Do Y, McKallip RJ, Nagarkatti M, et al. Activation through cannabinoid receptors 1 and 2 on dendritic cells trigger NF-κB-dependent apoptosis: Novel role for endgenous and exogenous cannabinoids in immunoregulation. J Immunol2004;173:2373–82.Abstract/FREE Full TextGoogle Scholar
- ↵ Qualtrough D, Buda A, Gaffield W, et al. Hedgehog signaling in colorectal tumour cells: induction of apoptosis with cyclopamine treatment. Int J Cancer2004;110:831–7.CrossRefPubMedWeb of ScienceGoogle Scholar
- ↵ Paglin S, Hollister T, Delohery T, et al. A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res2001;61:439–44.Abstract/FREE Full TextGoogle Scholar
- ↵ Gardner B, Zhu LX, Sharma S, et al. Methanandamide increases COX-2 expression and tumour growth in murine lung cancer. FASEB J2003;17:2157–9.Abstract/FREE Full TextGoogle Scholar
- Ramer R, Weinzierl U, Schwind B, et al. Ceramide is involved in R(+)-methanandamide-induced cyclooxygenase-2 expression in human neuroglioma cells. Mol Pharmacol2003;64:1189–98.Abstract/FREE Full TextGoogle Scholar
- ↵ Hinz B, Ramer R, Eichele K, et al. Upregulation of cyclooxygenase-2 expression is involved in R(+)-methanandamide-induced apoptotic death of human neuroglioma cells. Mol Pharmacol2004;66:1643–51.Abstract/FREE Full TextGoogle Scholar
- ↵ Deutsch DG, Omeir R, Arreaza G, et al. Methyl arachidonyl fluorophosphates: a potent irreversible inhibitor of anandamide amidase. Biochem Pharmacol1997;53:255–60.CrossRefPubMedWeb of ScienceGoogle Scholar
- ↵ Ross A, Craib SJ, Stevenson LA, et al. Pharmacological characterization of the anandamide cyclooxygenase metabolite: prostaglandin E2 ethanolamide. J Pharmacol Exp Ther2002;301:900–7.Abstract/FREE Full TextGoogle Scholar
- ↵ Mutoh M, Watanabe K, Kitamura T, et al. Involvement of prostaglandin E receptor subtype EP(4) in colon carcinogenesis. Cancer Res2002;62:28–32.Abstract/FREE Full TextGoogle Scholar
- ↵ Pozzi A, Yan X, Macias-Perez I, et al. Colon carcinoma cell growth is associated with prostaglandin E2/EP4 receptor-evoked ERK activation. J Biol Chem2004;279:29797–804.Abstract/FREE Full TextGoogle Scholar
- ↵ Movsesyan VA, Stoica BA, Yakovlev AG, et al. Anandamide-induced cell death in primary neuronal cultures: role of calpain and caspase pathway. Cell Death Diff2004;11:1121–32.CrossRefPubMedWeb of ScienceGoogle Scholar
- ↵ Kim J, Alger BE. Inhibition of cyclooxygenase-2 potentiates retrograde endocannabinoid effects in hippocampus. Nat Neurosci2004;7:697–8.CrossRefPubMedWeb of ScienceGoogle Scholar
- ↵ Fowler CJ, Jonsson KO, Andersson A, et al. Inhibition of C6 glioma cell proliferation by anandamide, 1-arachidonoylglycerol, and by water soluble phosphate ester of anandamide: variability in response and involvement of arachidonic acid. Biochem Pharmacol2003;66:757–67.CrossRefPubMedGoogle Scholar
- ↵ Butler R, Mitchell SH, Tindall DJ, et al. Non-apoptotic cell death associated with S-phase arrest of prostate cancer cells via the peroxisome proliferator-activated receptor γ ligand, 15-deoxy-Δ 12,14-prostaglandin J2. Cell Growth Diff2000;11:49–61.Abstract/FREE Full TextGoogle Scholar
- ↵ Okada H, Mak TW. Pathways of apoptotic and non-apoptotic death in tumour cells. Nature Rev Cancer2004;4:592–603.CrossRefPubMedWeb of ScienceGoogle Scholar
- ↵ Kozak KR, Prusakiewicz JJ, Rowlinson SW, et al. Amino acid determinants in cyclooxygenase-2 oxygenation of the endocannabinoid anandamide. Biochemistry2003;42:9041–90.CrossRefPubMedGoogle Scholar